Reports
Evolution in Management of Pulmonary Arterial Hypertension (PAH)
This report is based on medical evidence presented at sanctioned medical congress, from peer reviewed literature or opinion provided by a qualified healthcare practitioner. The consumption of the information contained within this report is intended for qualified Canadian healthcare practitioners only.
MEDICAL OPTIONS - Respirology
March 2016
Targeting of the Prostacyclin Pathway in PAH
Editorial Review
Written by: Theodore Bosworth
Steeve Provencher, MD, MPH, FRCPC
Director, Pulmonary Hypertension Program
Professor, Université Laval
Quebec City, Quebec
For the treatment of pulmonary arterial hypertension (PAH), intravenous (IV) and subcutaneous (SC) therapies acting on the prostacyclin pathway are typically considered only after patients on first-line oral agents have reached an advanced stage, usually functional class (FC) IV. With the development of novel oral therapies acting on the prostacyclin pathway, there is now an opportunity to address this pathway at an earlier point in disease progression.In the recently published phase 3 GRIPHON trial, the oral selective IP receptor agonist selexipag was associated with a significant reduction in the risk of a primary composite endpoint of death or a complication related to PAH.1 This article will discuss treatment options for PAH with the context of strategies that target the prostacyclin pathway.
Background
Pulmonary hypertension (PH) has been defined as a mean pulmonary arterial pressure ≥25 mmHg at rest on right heart catheterization.2 PAH, distinct from other forms of PH, has been defined as a pulmonary artery wedge pressure (PAWP) ≤15 mmHg and a pulmonary vascular resistance (PVR) >3 Wood units occurring in the absence of other underlying disorders.2 Although PAH can be associated with an array of conditions and causes, such as connective tissue disease, portal hypertension, and exposure to toxins, most cases are idiopathic.3
The estimates of the incidence and prevalence of PAH are likely to be inexact because of non-specific symptoms that complicate the effort to diagnose PAH. Consistent with proportional figures reported in Europe and the U.S., the number of cases of PH in Canada is believed to lie somewhere between 5000 to 15,000 of which PAH represents a substantial proportion.4,5 According to the Pulmonary Hypertension Association of Canada, it is not uncommon for patients to have symptoms of PAH for 2 to 3 years before obtaining an accurate diagnosis.5 Somewhat more common in women than men6 PAH can develop at any age, but the mean age at diagnosis in the United States and Europe has been reported recently to be in the range of 35 to 65 years of age.6-8
PAH is a chronic progressive disease that leads in its terminal stages to right ventricular failure. Prior to the development of targeted therapies for PAH, the median survival after diagnosis was less than 3 years.9 Incremental improvements in survival have followed the introduction of better treatment options and improved diagnosis so that 5-year survival rates now exceed 50%.10 Efforts to initiate and optimize therapy early in the course of PAH may offer the opportunity to slow disease progression with the ultimate goal of extending survival.11 Striving to maintain patients in a low risk status with optimal functional capacity is essential for this goal.
Prostacyclin Pathway
Prostacyclin (PGI2), which is released by endothelial cells, is one of five naturally-occurring prostanoids. A key mediator of interactive systems important to PAH progression, prostacyclin exerts its effects on biological function through membrane-bound cell surface receptors.12 Unlike some prostanoids, such as PGE2, which activates both EP1, a receptor associated with contractile activity, and EP2 and EP4, which are associated with relaxant activity, PGI2 activates only the IP receptor (see Figure 1). This receptor, associated with vasorelaxation, is found on vascular smooth muscle cells, epithelial cells, fibroblasts, and platelets. When activated, it not only upregulates cAMP, which is instrumental to vasorelaxation, but also triggers a downstream signaling cascade that includes antiproliferative effects, inhibition of platelet aggregation, and anti-inflammatory effects.13
Figure 1.
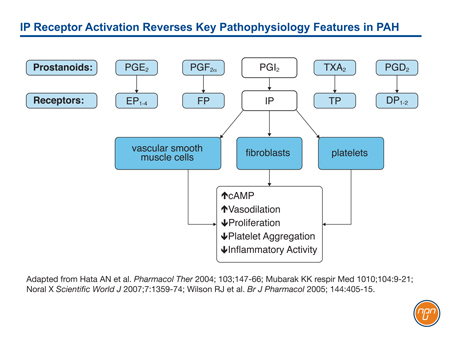
PAH is associated with both low levels of endogenous prostacyclin and suppressed prostacyclin activity,14 reinforcing the expected benefit of activating this pathway with epoprostenol, prostacyclin analogues, or novel therapies acting directly on prostacyclin receptors.
IV epoprostenol, a synthetic prostacyclin, became the first disease-specific therapy for PAH when it received regulatory approval in the mid 1990s. Interest in the prostacyclin pathway as target of therapy for PAH grew out of a series of experimental studies that followed the isolation of prostanoids in 1976.15 These studies culminated in a seminal multicentre clinical trial that associated epoprostenol with a survival benefit (P=0.003) relative to standard therapy in idiopathic PH patients in FC III or IV.16
Despite the anti-PAH activity of therapies that promote prostacyclin activity, these continue to be employed primarily in patients with advanced disease. This is due in large part to currently available formulations. Epoprostenol has a half-life measured in minutes,17 making continuous IV treatment the only feasible method of administration. Treprostinil, which may offer efficacy comparable to epoprostenol,18 is available in IV and SC formulations, but SC delivery by portable infusion pump is also relatively inconvenient. Moreover, local infusion site pain with SC delivery occurs in approximately 80% of patients.19
It is the successful development of oral agents acting to inhibit endothelin-mediated vasoconstriction or to promote nitric oxide (NO)- mediated vasodilation that have effectively relegated therapies acting on the prostacyclin pathway to treatment of advanced disease. Although oral and inhaled formulations of such prostacyclin analogues as treprostinil, iloprost, and beraprost have been developed, efficacy by these routes has been mixed. In the TRIUMPH study, which randomized 235 PAH patients in FC III or IV treated with bosentan or sildenafil were randomized to inhaled treprostinil or placebo.20 At week 12, the 6 minute walk distance (6MWD), quality of life and NT-proBNP were all improved on treprostinil relative to placebo. However, oral treprostinil failed to achieve a significant improvement relative to placebo in 6MWD when added to a stable background therapy of an endothelin-receptor antagonists (ERA) or phosphodiesterase-5 (PDE-5) inhibitor in either the FREEDOM-C study or the FREEDOM-C2 study.21,22 In an uncontrolled study with inhaled iloprost, only a minority of PAH patients were successfully stabilized in a long-term prospective evaluation.23
In Canada, the only options for treating PAH with agents acting on the prostacyclin pathway have been IV epoprostenol and IV and SC treprostinil. Acting on the other two targetable pathways, oral therapies include three ERAs, two PDE-5 inhibitors, and a soluble guanylate cyclase (sCG) inhibitor, which, like the PDE-5 inhibitors, targets the NO pathway. Due to relative convenience, oral agents are almost always employed initially. Often, several oral agents have been combined before PAH is considered sufficiently advanced to consider an IV or SC therapies acting on the prostacyclin pathway.
New Prostacyclin Pathway Strategies
Progress has been made in targeting the prostacyclin pathway with orally active agents that mediate the activity of prostacyclin receptors. These oral agents allow this mechanism of action to be introduced at a much earlier point in disease progression. Whether used alone or in combination with other oral therapies, agents able to promote the vasodilatory effects of prostacyclin at an early stage of disease have the potential to contribute to current efforts to slow progression of the underlying pathophysiology.
As the only functional relaxant prostacyclin receptor expressed in the pulmonary arteries,24 the IP prostacyclin receptor has been a logical and appropriate focus of drug development.25 IP is the key receptor activated by the prostacyclin prostanoid PGI2. Unlike several other receptor subtypes that have variable effects on vasodilation, smooth muscle cell proliferation, or both, IP has an important role in mediating vasodilatory and anti-proliferative effects.26 As opposed to selective IP receptor antagonists, prostaglandin analogues, such as iloprost and treprostinil, have variable affinity for a host of prostanoid receptors with varying activities, some of which would be expected to be counterproductive for treatment of PAH.13
Selexipag is the first oral selective IP prostacyclin-receptor agonist to reach advanced stages of development.27 In preclinical trials, selexipag demonstrated a high degree of affinity for the IP receptor,28 which mediates the vasodilatory effects of the prostacyclin pathway but low affinity for other prostacyclin receptors. A phase 2 proof-of-concept study confirmed a relatively modest although significant vasodilatory effect consistent with the activity observed in experimental studies.27 The phase 2 trial also confirmed the drug was well tolerated and suitable for further development.
In the phase 3 trial called GRIPHON,1 1156 PAH patients were randomized to selexipag or placebo. Patients on existing stable background therapy with an ERA, a PDE5 inhibitor, or both were eligible. Of those who entered the trial, 20% were treatment naïve, 47% were on one of these oral agents, and 33% were on both. The primary endpoint was a composite of death from any cause or a complication related to PAH up to the end of the treatment period. Complications were defined as disease progression (defined as a decrease from baseline of >15% in the 6-minute walk distance accompanied by a worsening in WHO FC or the need for additional treatment of PAH) or worsening of PAH that resulted in hospitalization, initiation of parenteral prostanoid therapy or long-term oxygen therapy, or the need for lung transplantation or balloon atrial septostomy.
The risk of the primary endpoint was reduced 40% in the arm treated with selexipag relative to placebo (HR 0.60, 95% CI, 0.46-0.78; P<0.001) (See Figure 2). The treatment effect was consistent across stratifications for age, gender, baseline FC, and background PAH therapy. The safety profile was consistent with other agents acting on the prostacyclin pathway. The most frequent adverse events included myalgias, flushing, headache, diarrhea and jaw pain.
Figure 2.

GRIPHON results, which confirm clinical benefit from an oral agent acting on the prostacyclin pathway, have already been incorporated in the joint 2015 guidelines from the European Society of Cardiology and European Respiratory Society (ESC/ERS) which conferred a level I recommendation for the addition of selexipag to an ERA agent, a PDE-5 inhibitor, or both in sequential combination therapy.11 This specific recommendation was made in the context of a more general recommendation to consider combination therapy in order to maintain patients in a low-risk status, particularly FC class II when possible. These updated PAH treatment recommendations reflect a reorientation toward improving long-term outcomes in addition to the immediate goal of improving quality of life.
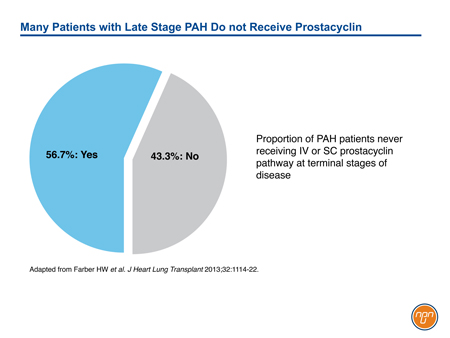
Although prostacyclin was the first pathway successfully targeted in the management of PAH, its role has been eclipsed by effective orally available treatments for the endothelin and NO pathways. It is noteworthy that IV prostanoid therapy remains the treatment of choice for high-risk patients, such as those in FC IV.11 However, even in advanced disease, therapies acting on the prostacyclin pathway appear to be underused. In a PAH registry capturing practice patterns in the United States, only 56.7% has received IV or SC prostacyclin or prostacyclin analogue within 6 months of death (see Figure 3).29 Progress in the development of novel oral agents targeting the prostacyclin pathway has important implications for expanding therapeutic options in PAH. Although long-term morbidity and mortality remains high in PAH, the GRIPHON trial documented that long-term treatment with an oral selective IP receptor agonist acting on the prostacyclin pathway is associated with reduced risk for complications related to PAH.
Figure 3.
Conclusion
According to registry data, survival in patients with PAH has improved in an era where therapies have been used earlier and in combination to slow disease progression.30 These combinations have relied primarily on oral ERA agents and PDE5 inhibitors. By demonstrating protection against clinical worsening, GRIPHON has provided evidence for introducing treatment active on the prostacyclin pathway at a relatively early stage of disease to improve outcomes. The effects of the oral agent selexipag appear to be independent and additive to those provided by oral therapies targeting the endothelin and NO pathways of PAH progression.
References
1. Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015;373:2522-33.
2. Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013;62:D42-50.
3. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004;351:1655-65.
4. Mehta SR. Living with PH. http://www.livingwithphca/landinghtm;December 1, 2015.
5. PHC. Pulmonary Hypertension Association of Canada. 2015.
6. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 2010;137:376-87.
7. Frost AE, Badesch DB, Barst RJ, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US Contemporary Registries. Chest 2011;139:128-37.
8. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173:1023-30.
9. Sandoval J, Bauerle O, Palomar A, et al. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation 1994;89:1733-44.
10. Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012;142:448-56.
11. "2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)." Nazzareno Galie, Marc Humbert, Jean-Luc Vachiery, et al. Eur Respir J 2015; 46: 903-975. Eur Respir J 2015;46:1855-6.
12. Zhang L, Huang G, Wu J, et al. A profile of the residues in the first intracellular loop critical for Gs-mediated signaling of human prostacyclin receptor characterized by an integrative approach of NMR-experiment and mutagenesis. Biochemistry 2005;44:11389-401.13.
13. Whittle BJ, Silverstein AM, Mottola DM, et al. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol 2012;84:68-75.
14. Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 1992;327:70-5.
15. Moncada S, Gryglewski R, Bunting S, et al. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 1976;263:663-5.
16. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301.
17. Safdar Z. Treatment of pulmonary arterial hypertension: the role of prostacyclin and prostaglandin analogs. Respir Med 2011;105:818-27.
18. McLaughlin VV, Gaine SP, Barst RJ, et al. Efficacy and safety of treprostinil: an epoprostenol analog for primary pulmonary hypertension. Journal Cardiovasc Pharmacol 2003;41:293-9.
19. Skoro-Sajer N, Lang I, Naeije R. Treprostinil for pulmonary hypertension. Vasc Health Risk Manag 2008;4:507-13.
20. McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010;55:1915-22.
21. Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): A randomized controlled trial. Chest 2012;142:1383-90.
22. Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest 2013;144:952-8.
23. Opitz CF, Wensel R, Winkler J, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J 2005;26:1895-902.
24. Wilson RJ, Giles H. Piglet saphenous vein contains multiple relaxatory prostanoid receptors: evidence for EP4, EP2, DP and IP receptor subtypes. Br J Pharmacol 2005;144:405-15.
25. Galie N, Ghofrani AH. New horizons in pulmonary arterial hypertension therapies. Eur Respir Rev 2013;22:503-14.
26 Norel X. Prostanoid receptors in the human vascular wall. ScientificWorldJournal 2007;7:1359-74.
27. Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J 2012;40:874-80.
28. Asaki T, Kuwano K, Morrison K, et al. Selexipag: An Oral and Selective IP Prostacyclin Receptor Agonist for the Treatment of Pulmonary Arterial Hypertension. J Med Chem 2015;58:7128-37.
29. Farber HW, Miller DP, Meltzer LA, et al. Treatment of patients with pulmonary arterial hypertension at the time of death or deterioration to functional class IV: insights from the REVEAL Registry. J Heart and Lung Transplant 2013;32:1114-22.
30. Pauwaa S, Machado RF, Desai AA. Survival in pulmonary arterial hypertension: A brief review of registry data. Pulmonary Circulation 2011;1:430-1.
Combination Therapy in PAH: Strategy to Delay Disease Progression
Editorial Review
Written by: Theodore Bosworth
John Swiston, MD, MPH, FRCPC
Division of Respiratory Medicine
University of British Columbia
Vancouver, BC
In the absence of a cure, the goal for the management of pulmonary arterial hypertension (PAH) is to slow progression. With the progressive advent of multiple medications that are individually effective in the treatment of PAH, the practice of combining treatments that employ different mechanisms of action becomes a logical strategy. Although this practice was first employed empirically, there is now a substantial body of evidence associating two or more therapies with protection from PAH-related clinical events relative to a single agent. This evidence includes several trials of new treatment options added to baseline therapy as well as a trial directly comparing a combination of two agents to either alone. It has not yet been established from these trials that early use of combination therapy alters the natural history of PAH, but preventing patients from advancing in functional class (FC) has been associated with better outcomes. Current guidelines support combination therapy as appropriate strategy, and registry data show that the majority of PAH patients are now being managed on two or more therapies.
Background
There are three major molecular pathways targeted for the treatment of PAH (see Figure 1). Each of these pathways, prostacyclin, endothelin, and nitric oxide (NO), is implicated in signaling cascades that modulate the imbalance of vasoconstriction/vasodilation, vascular cell proliferation, and inflammation that characterizes progressive PAH. Each is associated with clinical benefit when targeted independently, but additive benefit may be achieved when two or all three are targeted simultaneously.
Figure 1.
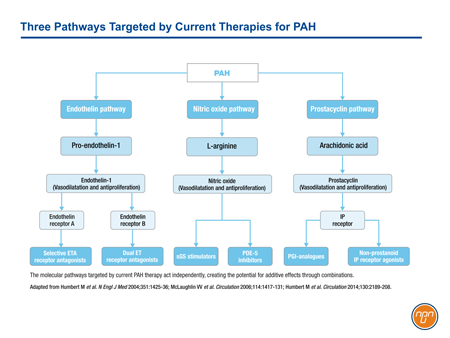
Prostacyclin was the first of three major pathways mediating PAH to be targeted clinically. The activity of prostacyclin, a potent endogenous vasodilator released by endothelial cells, is impaired in PAH.1 Receptors activated in the pulmonary circulation, such as those on smooth muscle cells, trigger signals that mediate vasorelaxation as well as anti-proliferative and anti-inflammatory effects.2 The first agent approved for the targeting of the prostacyclin pathway, intravenous (IV) epoprostenol, significantly improved 6-minute walk distance (6MWD) at the end of 12 weeks in patients with FC III or IV PAH.3 Other agents acting on the prostacyclin pathway followed.
The endothelin pathway was the second to be successfully targeted in the treatment of PAH. Endothelin (ET-1) is a potent vasoconstrictor that triggers signaling important to interstitial remodeling characteristic of PAH.4 ET-1 levels have been shown to be elevated in patients with PAH relative to those with normal lung function.5 The first endothelin-receptor antagonist (ERA) licensed for the treatment of PAH, bosentan, also provided an improvement in 6MWD relative to placebo.6 Subsequent studies led to the approval of ambrisentan and, in a longer trial using a rigorous composite morbidity and mortality endpoint, macitentan.7,8
The third pathway involves strategies to enhance nitric oxide (NO)-mediated vasodilation. Sildenafil, the first agent approved for PAH to target this pathway inhibits phosphodiesterase-5 (PDE-5), an enzyme that breaks down and deactivates the second messenger, cyclic guanosine monophosphate (cGMP).9 In the pivotal phase 3 trial, sildenafil improved 6MWD at the end of 12 weeks relative to placebo.10 Tadalafil, another PDE-5 inhibitor available for PAH has also shown sustained improvements in 6MWD.11 Riociguat, the most recently approved therapy to target the NO pathway, directly stimulates soluble guanosine cyclase (sCG) leading to increased production of cGMP.12 It, too, was associated with improvements in 6MWD at the end of 16 weeks on therapy.13
From an initial goal of providing short-term functional improvements, the reorientation toward improving long-term outcomes in PAH is reflected in current trial endpoints. All three treatment pathways of PAH are linked to inhibition of the underlying cascade of pathophysiologic events that includes inflammation, fibrosis, and vascular remodeling.14-16 Earlier diagnosis of PAH and more intensive blockade of these events may be at least partially responsible for improving PAH survival, which climbed from approximately 3 years in 1990 prior to the advent of PAH-specific treatment to 7 years in 2012.17
Combination Therapy
The combination of therapies targeting distinct pathways of PAH is an intuitive strategy. Although not all of the many small studies evaluating combination therapy demonstrated a significant advantage for adding a second agent to monotherapy,18 several larger studies, such as PACES,19 which showed an advantage for time to clinical worsening for the addition of oral sildenafil relative to IV epoprostenol alone, have supported this approach.20 A meta-analysis published in 2012 concluded that combination therapies improve both hemodynamics and clinical outcomes.21 By the time of publication of this meta-analysis, a large PAH registry in the United States revealed that almost half of PAH patients were being managed on two medications and nearly 10% were managed on three.22
Recent clinical trials have contributed substantially to evidence that combination therapy is appropriate in PAH patients who are not adequately managed on a single drug. This not only includes trials specifically designed to test combination therapy but also placebo-controlled trials of newer agents that enroll treatment-experienced patients on background therapy. By including patients on background therapy, these trials evaluate a subset of patients taking the experimental agent in combination.
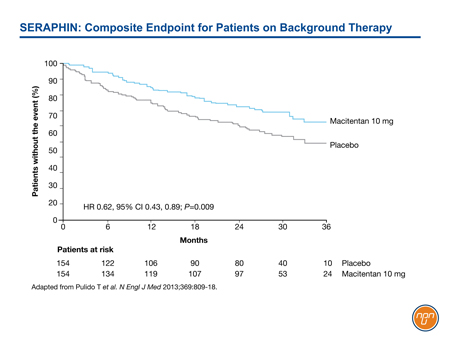
In SERAPHIN, for example, 64% of enrolled patients were taking and remained on background therapy, primarily a PDE-5 inhibitor.8 Despite the large proportion of patients on an active agent, the 10-mg dose of macitentan provided a 45% reduction (HR 0.55, 95% CI 0.39, 0.76; P<0.001) relative to placebo in the risk of the composite endpoint of death, atrial septostomy, lung transplantation, initiation of treatment with IV or SC prostanoids, or worsening or PAH. Among those on background therapy, the 38% reduction in the risk of the primary composite endpoint remained statistically significant (HR 0.62, 95% CI 0.43, 0.89; P=0.009) (see Figure 2).
Figure 2.
In the PATENT-1 study, testing the sGC agonist riociguat, approximately 50% of the study population was enrolled on a background agent, primarily an ERA agent.23 The primary endpoint in this study was change in 6MWD at 12 weeks. Although this may be the last major PAH trial not to include morbidity/mortality events as the primary evaluation of benefit, this study also showed an advantage for the novel agent relative to placebo 6MWD despite the substantial proportion of individuals already taking an active therapy.
The recently completed GRIPHON study, which is the largest clinical trial ever conducted in PAH, evaluated the novel selective IP prostacyclin receptor agonist selexipag in 1156 patients.24 In this placebo-controlled trial only 20% of participants were treatment naïve. While nearly 50% were taking either a PDE-5 inhibitor or an ERA agent, 33% were taking both. Despite the high proportion of patients on up to 2 active agents, the addition of selexipag was associated with a 40% reduction (HR 0.60, 95% CI 0.46, 0.78; P<0.001) in morbidity/mortality events. These data are consistent with a small study associating a combination of three drugs acting on each of the targetable pathways of PAH with long-term benefits in advanced PAH,25 but GRIPHON is the first large prospective trial of this strategy. Moreover, the efficacy of targeting all 3 pathways was observed in patients at an earlier stage of PAH. Of those randomized 44% were in FC II at enrollment.
The AMBITION trial was also completed recently. This trial was designed specifically to test a combination relative to a single-agent as first-line treatment.26 Enrollment was limited to patients who were treatment naïve. Like SERAPHIN and GRIPHON, AMBITION employed a primary composite morbidity/mortality endpoint. Patients were randomized to ambrisentan plus placebo, tadalafil plus placebo, or a combination of the two active agents. In those randomized to the combination, there was a 50% reduction in morbidity and mortality (HR 0.50, 95% CI 0.35, 0.72; P<0.001) relative to those receiving a single active therapy.
Overall, recent trials have generated a large increase in the quantity of data that support combination therapy to improve outcomes in PAH. Although the evidence of benefit from a second agent in trials that permitted background therapy is indirect, the protection against clinical worsening in patients already on an active therapy is consistent with an expectation of added benefit from targeting two disease pathways.
Relative to short-term functional improvements, the composite morbidity/mortality endpoints now employed in major clinical trials have focused the goal of treatment in PAH on improvement in long-term outcomes. Building on the evidence of benefit from combination therapy observed in SERAPHIN and AMBITION, GRIPHON was the first randomized controlled trial to include a substantial proportion of patients on triple therapy. These data emphasize the value of targeting several therapeutic pathways relatively early in disease with the objective of improving outcomes.
Guidelines: Role of Combinations
The 2015 joint guidelines from the European Society of Cardiology and the European Respiratory Society (ESC/ERS) accept combination therapy, including the use of triple therapy, with the goal or maintaining or returning patients to FC class II.27 Although previous guidelines have endorsed combination strategies as acceptable in patients with advancing PAH, the ESC/ERS guidelines are the first to conclude that this is an evidence-based recommendation. In these guidelines, combination therapy is recommended even for patients in FC class II when required to maintain patients at a low risk status. Based on published studies, three strategies were identified as meeting class I level of evidence for a sequential combination.
In a meta-analysis cited in the ESC/ERS guidelines, combination therapy was associated with a 52% relative reduction (RR 0.48, 95% CI 0.26, 0.91, P=0.023) in the risk of clinical worsening relative to controls with no increase in the rate of serious adverse events.15
In outlining both upfront combination and sequential-agent combination therapy as viable strategies for the treatment of PAH, the authors of the ESC/ERS guidelines acknowledge the importance of combining mechanisms of action when targets cannot be reached on a single drug (see Figure 3). The three sequential combinations with a level I evidence of benefit for FC II PAH are macitentan added to sildenafil, selexipag added to an ERA agent and/or a PDE-5 inhibitor, and riociguat added to bosentan. The same combinations plus sildenafil added to epoprostenol were considered to have level I evidence for FC III. The GRIPHON study with selexipag provides the best prospective evidence of benefit from combining agents active against all three current targets in PAH.
Figure 3.
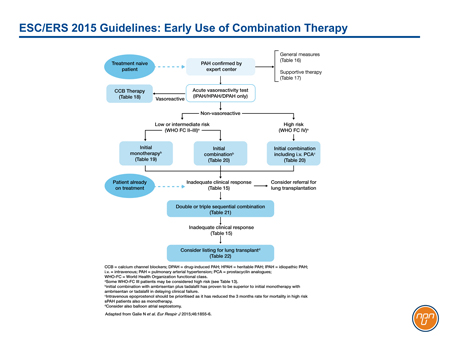
Better outcomes have been associated with preventing patients from advancing in FC.28 While previous guidelines have acknowledged the potential benefits of combination therapy, the most recent ESC/ERS recommendations provide an evidence basis for this approach. The guidelines suggest that the decision to add therapies in PAH may be reasonable even in patients who are stable or have improved slightly but have not yet reached treatment goals, such as an improvement in FC.
Conclusion
The current ESC/ERS guidelines advocate combining therapies in order to maintain PAH patients in a low-risk status. This recommendation is based on multiple trials demonstrating benefit when distinct treatment pathways are targeted simultaneously. In the guidelines, combining drugs acting on all three pathways may be considered as early as FC II when needed to sustain a low-risk status, which has been associated with an improved prognosis. This specific recommendation, supported by the recent GRIPHON study, establish a trajectory that has taken the treatment of PAH from monotherapy to dual combination therapy and now on to triple combination therapy in an effort to slow disease progression even in early stages of disease.
References
1. Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999;159:1925-32.
2. Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
3. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301.
4. Dupuis J, Hoeper MM. Endothelin recepotr antagonists in pulmonary arterial hypertension. Eur J Respir 2008;31:407-15.
5. Rubens C, Ewert R, Halank M, et al. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest 2001;120:1562-9.
6. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903.
7. Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010-9.
8. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18.
9. Barnett CF, Machado RF. Sildenafil in the treatment of pulmonary hypertension. Vasc Health Risk Manag 2006;2:411-22.
10. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148-57.
11. Oudiz RJ, Brundage BH, Galie N, et al. Tadalafil for the treatment of pulmonary arterial hypertension: a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol 2012;60:768-74.
12. Grimminger F, Weimann G, Frey R, et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J 2009;33:785-92.
13. Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369:319-29.
14. Fukumoto Y, Shimokawa H. Recent progress in the management of pulmonary hypertension. Circ J 2011;75:1801-10.
15. Galie N, Palazzini M, Manes A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J 2010;31:2080-6.
16. Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010;121:2045-66.
17. Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012;142:448-56.
18. Ruiz G, Besinque GM, Lickert CA, et al. Combination therapy in pulmonary arterial hypertension: is this the new standard of care? Am J Manag Care 2015;21:s151-61.
19. Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521-30.
20. Ghofrani HA, Humbert M. The role of combination therapy in managing pulmonary arterial hypertension. Eur Respir Rev 2014;23:469-75.
21. Zhu B, Wang L, Sun L, et al. Combination therapy improves exercise capacity and reduces risk of clinical worsening in patients with pulmonary arterial hypertension: a meta-analysis. J Cardiovasc Pharmacol 2012;60:342-6.
22. McGoon MD, Miller DP. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur Respir Rev 2012;21:8-18.
23. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330-40.
24. Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015;373:2522-33.
25. Sitbon O, Jais X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J 2014;43:1691-7.
26. Galie N, Barbera JA, Frost AE, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med 2015;373:834-44.
27. "2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)." Nazzareno Galie, Marc Humbert, Jean-Luc Vachiery, et al. Eur Respir J 2015; 46: 903-975. Eur Respir J 2015;46:1855-6.
28. Nickel N, Golpon H, Greer M, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2012;39:589-96.
Achieving Long-term Goals in PAH Therapy
Editorial Review
Written by: Theodore Bosworth
Ali M. Kapasi, MD, FRCPC
Division of Pulmonary Medicine
University of Alberta
Edmonton, Alberta
Pulmonary arterial hypertension (PAH) is driven by complex pathophysiology that includes vasoconstriction, cell proliferation, fibrosis and in-situ thrombosis. The progressive impairment in hemodynamics driven by this process ultimately results in right-heart failure. Initially, therapies targeted at PAH were evaluated based on their ability to achieve short-term improvements in functional capacity, such as change in 6-minute walk distance (6MWD) over a period of 3 or 4 months. As the field has evolved, and as more pharmacologic options have become available, the focus has shifted to attenuation of long-term disease progression. This shift is reflected in the endpoints of recent clinical trials, which now assess treatment impact on composite endpoints encompassing morbidity and mortality, including worsening of PAH, hospitalization, transplant and death. These trials suggest that intensified treatment can slow disease progression. In the past, worsening symptoms or a decline in functional capacity have been the primary impetus to intensify therapy, but the most recently published European Cardiology Society – European Respiratory Society (ESC/ERS) guidelines recommend adjusting treatment to achieve target goals that aim to maintain patients at low risk status for disease progression.
Background
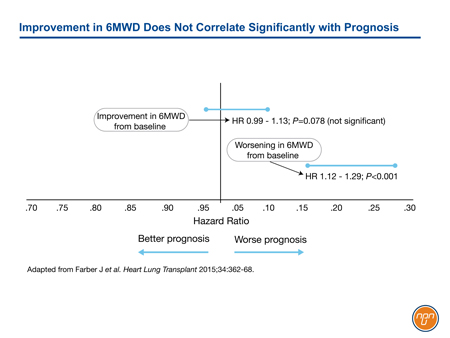
The goals of treatment in PAH, a progressive disease with a poor prognosis, were initially modest. Regulatory approval of intravenous (IV) epoprostenol, the first therapy approved specifically for PAH, was based on a trial that employed a primary endpoint of improvement in the 6MWD at the end of 12 weeks.1 This test of efficacy was reasonable at a time when many patients with PAH were diagnosed at an advanced stage and when median survival was only three years. The U.S. Food and Drug Administration (FDA) subsequently mandated this outcome as a test of benefit. This same endpoint continued to be used in many subsequent pivotal trials, including those for oral bosentan,2 subcutaneous (SC) treprostinil,3 and oral sildenafil.4 Consistent with several other studies, data from the REVEAL registry has now demonstrated that although worsening of 6MWD predicts a poor prognosis, improvement in 6MWD does not correlate with improved survival (see Figure 1).5 In this study, no 6MWD threshold was found to have prognostic value.
Figure 1.
An expert consensus group first recommended morbidity and mortality endpoints for clinical trials in 2009.6 The goal of this recommendation was to highlight potential differences between therapies in terms of their ability to change longer term outcomes. SERAPHIN, completed in 2013 was the first multicentre, randomized, trial to employ a composite of morbidity and mortality events as the primary endpoint.7 This endpoint was defined as the time from the initiation of treatment to the first occurrence of worsening of PAH, initiation of treatment with IV or SC prostanoids, atrial septostomy, lung transplantation, or death. Relative to placebo, the 10 mg dose of the endothelin-receptor antagonist (ERA) macitentan produced a 45% reduction (P<0.001) in this composite endpoint. Several subsequent PAH trials have also employed clinical events rather than functional improvements alone as the primary endpoint to evaluate treatment benefits.
As the disease progresses, PAH is characterized by a vicious cycle of increased vascular resistance, cell proliferation, and vascular remodeling.8 The greatest opportunity for benefit from treatment of PAH is likely to occur before tissue damage becomes irreversible. For intravenous (IV) epoprostenol, larger survival benefits were seen among patients in lower relative to higher functional classes (FC).9 Similarly, the greatest opportunity for benefit from other therapies, including oral agents, are likely to be achieved when they are still able to attenuate vascular remodeling and other pathophysiologic changes.
In addition to symptom relief and functional improvements,10 new joint treatment guidelines from the ESC/ERS suggest prognostic markers as treatment targets; changing or intensifying therapy with the goal of reaching these treatment targets to ultimately slow disease progression. Such prognostic markers include normalization of resting hemodynamics, normalization of plasma levels of N-terminal pro b-type natriuretic peptide (NT-proBNP), and maintenance of VO2 max > 15 mL/kg/min.
Selecting Initial Therapy
The treatment of PAH has evolved along three distinct pathways. Initial therapies for PAH targeted the prostacyclin pathway, which is associated with vasodilatory, anti-inflammatory, anti-proliferative, and anti-thrombotic effects.11 The second class of agents introduced for PAH targeted the endothelin (ET) pathway. Two receptor types, ETA and ETB, mediate the effects of ET-1, which is the predominant ET isoform. ET-1 induces vasoconstriction, fibroblast proliferation, and vascular remodeling.12,13 Nitric oxide (NO), a potent vasodilator and regulator of smooth muscle proliferation with potent signaling of other changes relevant to PAH, such as platelet aggregation,14 is the third targetable pathway. The NO pathway can be targeted through inhibition of phosphodiesterase-5 (PDE-5) or stimulation of soluble guanylate cyclase (sGC).4,15
Epoprostenol, which targets the prostacyclin pathway, is the only targeted treatment that has been associated with a survival benefit in a clinical trial.1 This trial was conducted in patients with highly advanced IPAH. The lower mortality in the epoprostenol arm was observed after only 12 weeks of therapy. Unfortunately, treatment with epoprostenol is relatively cumbersome for patients as it requires continuous IV infusion. As such, epoprostenol is generally reserved for patients with severe (FC IV) or refractory disease.
More recently, the trend has been towards recruitment of patients in FC II or III into clinical trials. When therapies are introduced at an earlier stage, mortality benefits have generally not been demonstrable over the course of clinical trials, thus making composite morbidity-mortality end-points more important and relevant. In trials such as the SERAPHIN trial with macitentan and the AMBITION trial with ambrisentan plus tadalafil, nearly all patients were in FC class III or lower. In these two studies, the proportions in FC class II were 45% and 30%, respectively. In each of these studies, macitentan and the combination of ambrisentan/tadalafil produced a significant reduction in their composite morbidity/mortality endpoints.7,16
Another recent trend in clinical trials has been the evaluation of new PAH therapies in both treatment naïve patients and those on stable background PAH treatment. In PATENT-1, which evaluated the sCG stimulator riociguat, approximately 50% of patients enrolled were on background therapy.17 In SERAPHIN, more than 60% of patients were on background therapy. In GRIPHON, the most recent and largest trial yet conducted in PAH, that proportion was 80%. In fact, 33% of those enrolled in GRIPHON were taking both an ERA agent and a PDE-5 inhibitor. In this study evaluating selexipag, an orally available selective IP receptor agonist targeting the prostacyclin pathway, a 40% reduction in a composite morbidity-mortality endpoint was seen.18
By demonstrating protection against disease progression over an extended period of follow-up, these trials support goals that are more robust than earlier studies. In accordance, PAH treatment guidelines have been adjusted. In addition to functional improvement, the 2015 ESC/ERS treatment recommendations place new emphasis on treatment titration with the objective of minimizing the risk of clinical deterioration.
Sildenafil, tadalafil, ambrisentan, bosentan, macitentan, riociguat and selexipag all received class I recommendations as potential initial choices for monotherapy in patients with PAH who are in FC II or III. Epoprostenol received a class I recommendation as initial monotherapy for PAH patients in FC III or IV, and SC treprostinil received a class I recommendation for PAH patients in FC III (as did inhaled treprostinil and inhaled iloprost, however these agents are not presently available in Canada). The combination of ambrisentan and tadalafil received a class I recommendation for initial combination therapy in PAH patients in FC II or III. As a consequence, there is a broad range of options to choose from for patients in FC II or III including either initial monotherapy with one of a variety of agents or initial combination therapy. Given the lack of head-to-head comparisons amongst these various options, it is difficult to make an across-the-board evidence-based recommendation for first-line therapy. It also remains unknown whether initial combination therapy offers additional benefits over sequential combination therapy. As such, the decision on which option to choose is based on several factors including patient preference, physician experience, side-effect profile, potential for interaction with a patient’s other medications and the individual patient’s co-morbidities. The one exception to this is patients presenting in FC IV, where IV epoprostenol is still the preferred option.
Stepping Up Therapy: Clinical Markers
The evidence that morbidity/mortality events can be reduced with current therapies has validated a shift in treatment goals. Recent trials, including those studies that have tested new treatment options on top of stable background regimens, suggest that intensification of treatment is an appropriate strategy to slow disease progression. The ESC/ERS guidelines provide a model for goal-oriented therapy in which the decision to modify therapy (including the use of combination therapy) is based on the objective of minimizing the risk of future deterioration.10 An emphasis is placed on maintaining or returning patients to FC II whenever possible. A second focus is placed on titration of therapy to achieve clinical markers of low-risk status. These markers include optimization cardiac index (CI), N-terminal pro b-type natriuretic peptide (NT-proBNP) and maximum oxygen consumption (VO2 max).19
In the ESC/ERS guidelines, both upfront and sequential combination strategies are considered appropriate for sustaining PAH patients in a low-risk state (see Figure 2). Because of their expected synergistic effects, the thinking is that multiple agents with demonstrated long-term outcome benefits can be combined in a step-wise fashion so that each of the known pathways leading to the pathogenesis of PAH are simultaneously targeted.
Figure 2.
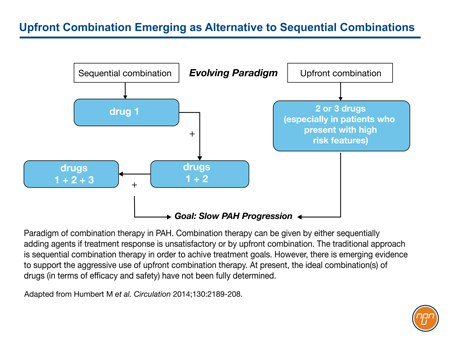
With previous iterations of the ESC/ERS guidelines, treatments acting on the prostacyclin pathway were typically reserved for advanced disease. In GRIPHON, approximately 45% of enrolled patients were in FC II. The benefit of the IP prostacyclin receptor agonist selexipag was demonstrated on top of background therapy, which included an ERA agent, a PDE-5 inhibitor or both in 80% of the study population (or both in 33% of patients). This trial appears to support early introduction of an agent acting on the prostacyclin pathway, even in patients who are stable on background therapy with a PDE-5 inhibitor and/or an ERA agent. In the ESC/ERS guidelines, risk has been characterized as low, intermediate, or high based on established prognostic factors (see Table 1).
Table 1.
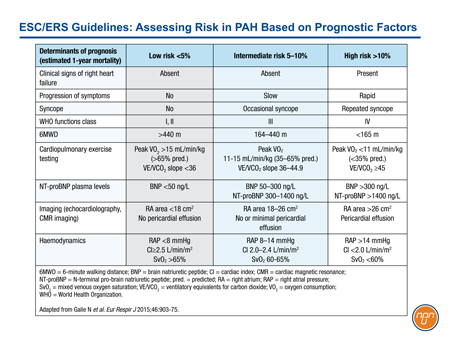
Conclusion
Treatment goals in PAH have evolved to reflect recent evidence that current therapies can reduce morbidity and are likely more effective in combination. In the recently issued ESC/ERS guidelines, focus has been placed on monitoring prognostic indicators and adjusting therapy to prevent clinical deterioration. An emphasis on long-term outcomes is clearly reflected in these guidelines.
The evolution of care in PAH is the product of many advances over the past two decades. Efforts to improve early recognition of PAH allow treatment to be initiated at earlier stages. Event-driven trials have demonstrated that available treatments are capable of achieving treatment goals beyond symptom improvement. Oral drugs targeting each of the known pathophysiologic mechanisms of PAH may permit earlier intensification of therapy, potentially delaying the development of additional morbidity and mortality. Parenteral drugs remain good options for patients with severe disease or disease which is refractory to oral therapy.
References
1. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301.
2. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903.
3. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165:800-4.
4. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148-57.
5. Farber HW, Miller DP, McGoon MD, et al. Predicting outcomes in pulmonary arterial hypertension based on the 6-minute walk distance. J Heart Lung Transplant 2015;34:362-8.
6. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009;119:2250-94.
7. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18.
8. Sakao S, Tatsumi K, Voelkel NF. Reversible or irreversible remodeling in pulmonary arterial hypertension. American Journal of Respiratory Cell and Molecular Biology 2010;43:629-34.
9. Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002;40:780-8.
10. "2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)." Nazzareno Galie, Marc Humbert, Jean-Luc Vachiery, et al. Eur Respir J 2015; 46: 903-975. Eur Respir J 2015;46:1855-6.
11. Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J 2008;31:891-901.
12. Dupuis J, Hoeper MM. Endothelin recepotr antagonists in pulmonary arterial hypertension. Eur J Respir 2008;31:407-15.
13. Sitbon O, Morrell N. Pathways in pulmonary arterial hypertension: the future is here. Eur Respir Rev 2012;21:321-7.
14. Zuckerbraun BS, George P, Gladwin MT. Nitrite in pulmonary arterial hypertension: therapeutic avenues in the setting of dysregulated arginine/nitric oxide synthase signalling. Cardiovasc Res 2011;89:542-52.
15. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330-40.
16. Galie N, Barbera JA, Frost AE, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med 2015;373:834-44.
17. Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369:319-29.
18. Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015;373:2522-33.
19. Nickel N, Golpon H, Greer M, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2012;39:589-96.