Comptes rendus
Points de vue sur l’hypertension artérielle pulmonaire (HTAP) - Revues de la littérature
Le présent compte rendu est fondé sur des données médicales présentées lors d'un congrès de médecine reconnu ou publiées dans une revue avec comité de lecture ou dans un commentaire signé par un professionnel de la santé reconnu. La matière abordée dans ce compte rendu s'adresse uniquement aux professionnels de la santé reconnus du Canada.
OPTIONS MÉDICALES - Pneumologie
Février 2014
Test de marche de 6 minutes : les paramètres d’évaluation de l’HTAP revisités
Commentaire éditorial
Rédaction : Theodore Bosworth
Paul Hernandez, MDCM, FRCPC
Pneumologue, QEII Health Sciences Centre
Professeur titulaire, Département de médecine
Dalhousie University
Halifax (Nouvelle-Écosse)
Largement utilisé, peu coûteux et facile d’utilisation en ambulatoire, le test de marche de 6 minutes (TM6M) mesure la capacité d’exercice fonctionnelle. Dans l’hypertension artérielle pulmonaire (HTAP), le médecin peut s’en servir en cabinet à la fois pour évaluer l’état fonctionnel de son patient et pour surveiller sa réponse au traitement. En outre, comme le TM6M est le paramètre principal des essais cliniques sur l’HTAP depuis une vingtaine d’années, il est à la base de l’homologation de la plupart des agents actuellement sur le marché1. Dans ces essais comparatifs avec placebo, le TM6M mesure objectivement la variation de la capacité fonctionnelle. Par contre, il n’a jamais été démontré que la variation de la distance parcourue au TM6M était un prédicteur sensible de l’issue à long terme, ce qui constitue une lacune importante lorsqu’on compare deux traitements actifs capables de ralentir la progression de la maladie. C’est précisément à cause de cette lacune que d’autres paramètres, surtout la survie ou un paramètre mixte incluant la survie, se substituent au TM6M à titre de paramètre principal d’évaluation du bénéfice associé aux nouvelles stratégies thérapeutiques.
Contexte : le TM6M dans la pratique clinique
Les épreuves d’effort réalisées en ambulatoire et en laboratoire sont des outils pratiques d’évaluation de la capacité fonctionnelle. Il existe divers tests, mais le TM6M – qui mesure la distance parcourue en 6 minutes sur une surface dure sans appareil particulier – est révélateur de la capacité à accomplir les activités de la vie quotidienne (AVQ)2. En pneumologie, il est souvent utilisé pour l’évaluation clinique de l’hypertension artérielle pulmonaire (HTAP), de la maladie pulmonaire obstructive chronique (MPOC), des maladies pulmonaires interstitielles (MPI) et d’autres maladies se caractérisant par une atteinte modérée ou sévère de la fonction pulmonaire3.
Dans ses recommandations de 2002 pour l’évaluation pratique de l’HTAP en milieu clinique (Tableau 1), l’American Thoracic Society (ATS) précise que le TM6M est un meilleur baromètre de la variation de la qualité de vie et de la capacité d’un patient à accomplir ses AVQ que certains paramètres de la fonction pulmonaire comme la consommation maximale d’oxygène3. L’ATS émet toutefois une réserve quant aux maigres données étayant la distance parcourue au TM6M comme paramètre unique d’évaluation de l’état fonctionnel ou comme prédicteur de l’issue clinique à long terme chez le patient atteint d’une maladie respiratoire.

Plus de 10 ans après la publication du guide de pratique de l’ATS, l’utilité de la distance parcourue au TM6M en tant que prédicteur de l’issue clinique à long terme ou marqueur de substitution d’un changement dans l’évolution naturelle de la maladie, et non comme paramètre d’évaluation du soulagement des symptômes, demeure controversée. Dans la MPOC, par exemple, les seuils de distance parcourue au TM6M permettant de prédire la mortalité sur une période de 3 ans variaient selon les caractéristiques du patient, l’âge en particulier4. On a aussi remis en question la sensibilité de la variation de la distance parcourue au TM6M pour l’évaluation du bénéfice associé à la bronchodilatation dans la MPOC5. Dans les MPI associées à la sclérodermie, l’impossibilité d’utiliser le TM6M pour prédire l’issue clinique de la composante respiratoire a été attribuée à d’autres atteintes physiques faisant obstacle à la capacité fonctionnelle6.
Compte tenu de la prolifération de nouveaux traitements qui, utilisés seuls ou en association, peuvent ralentir la progression de la maladie, il est devenu impératif de déterminer si le TM6M est un prédicteur utile de l’issue clinique dans l’HTAP. Le soulagement des symptômes est important, certes, mais comme les traitements peuvent infléchir l’évolution naturelle de cette maladie, il est essentiel de comparer leurs effets respectifs afin de définir la prise en charge optimale.
Mesure du fardeau clinique de l’HTAP
Le soulagement des symptômes que procure le traitement de l’HTAP – maladie complexe dont les étiologies sont multiples et dont les sous-types sont cliniquement pertinents7,8 – n’est pas forcément corrélé avec une protection contre la morbidité et la mortalité associées aux complications systémiques de la maladie. En raison du vaste éventail de changements structuraux, mécaniques et biochimiques qu’elle entraîne, beaucoup de variables pourraient être utiles pour le dépistage et la surveillance de l’HTAP7. Il importe de reconnaître que, parmi les variables cliniquement pertinentes, certaines pourraient être corrélées avec l’expression des symptômes, d’autres avec la progression de la physiopathologie sous-jacente et d’autres encore, avec les deux.
Sur le plan clinique, l’amélioration de la classe fonctionnelle – ce qui sous-entend une diminution des symptômes – est un paramètre valable pour l’évaluation du bénéfice associé au traitement même lorsque ces symptômes ne peuvent pas être considérés comme des marqueurs de substitution d’une meilleure issue clinique. Les classes fonctionnelles de l’Organisation mondiale de la Santé (OMS) – qui vont de I (aucune conséquence des symptômes sur l’activité clinique) à IV (présence de symptômes de l’HTAP, comme la dyspnée, au repos) – témoignent déjà du fardeau clinique de l’HTAP, et sont corrélées avec la mortalité9; les données montrant qu’une amélioration de la classe fonctionnelle se traduit directement par une prolongation de la survie sont toutefois moins concluantes. De même, la capacité d’exercice évaluée par des outils à la fine pointe de la technologie comme l’épreuve d’effort cardio-pulmonaire (CPET), qui mesure les échanges gazeux métaboliques au repos et à l’effort, a aussi une valeur pronostique10, quoique le lien entre l’amélioration attribuable au traitement et l’amélioration de l’issue clinique à long terme demeure incertain.
Dans notre quête d’une évaluation du bénéfice associé au traitement de l’HTAP, nous devrions tenir compte à la fois de paramètres cliniques majeurs – à savoir, une réduction objective des hospitalisations et, en définitive, de la mortalité – et de paramètres subjectifs moins tangibles comme la qualité de vie. Les deux sont ciblés dans le débat sur la sélection optimale des paramètres d’un essai clinique sur l’HTAP. Une diminution des symptômes permettant aux patients de vaquer à leurs occupations quotidiennes est bien sûr un objectif important du traitement, mais un traitement optimal doit à la fois atténuer les symptômes et ralentir la progression de la maladie, bénéfice qui se présente sous forme d’une incidence moindre d’événements majeurs.
Le TM6M : paramètre de substitution dans l’HTAP
Plusieurs considérations pratiques expliquent que la distance parcourue au TM6M ait été le paramètre principal des premiers essais sur les traitements médicamenteux, notamment la difficulté de faire ressortir un gain de survie sur une période de généralement 12 à 24 semaines seulement11, mais on n’a jamais réussi à démontrer que c’était un paramètre de substitution valable pour l’évaluation d’un changement dans la progression de la maladie. Comment, donc, expliquer que la distance parcourue au TM6M soit demeurée le paramètre principal des essais cliniques? Question de précédent, peut-être, car les essais d’envergure reposent typiquement sur beaucoup d’autres variables biologiques, radiologiques et cliniques pour faire ressortir une amélioration des fonctions pulmonaire et cardiaque. Plusieurs groupes arguent depuis quelques années déjà que le paramètre principal des essais sur l’HTAP devrait inclure la survie, car il n’y a probablement pas de meilleure preuve de la supériorité d’un traitement par rapport à un autre11-13. En parallèle, il ressort d’un nombre croissant de données, y compris les analyses de l’essai SERAPHIN qui vient de se terminer, que la variation de la distance parcourue au TM6M n’est peut-être pas un prédicteur exact de la survie.
Une méta-analyse de 22 essais avec randomisation opposant des agents actifs et un placebo a objectivé une réduction significative de la mortalité toutes causes confondues (p<0,01) de même qu’une réduction significative des hospitalisations pour cause d’HTAP et des transplantations pulmonaires (paramètre mixte) (p<0,01), mais elle n’a fait ressortir aucun lien significatif entre la distance parcourue au TM6M et les hospitalisations ou la mortalité toutes causes confondues (Figures 1a et 1b)14. Une autre méta-analyse de 10 essais a révélé que le traitement actif était associé à une diminution de plus de 50 % du risque relatif approché (OR [odds ratio]) d’événement clinique, mais une méta-régression a permis de constater que la variation de la distance parcourue au TM6M expliquait seulement 22 % de cet effet du traitement et constituait un paramètre de substitution médiocre pour la détection d’une diminution de l’incidence des événements15.
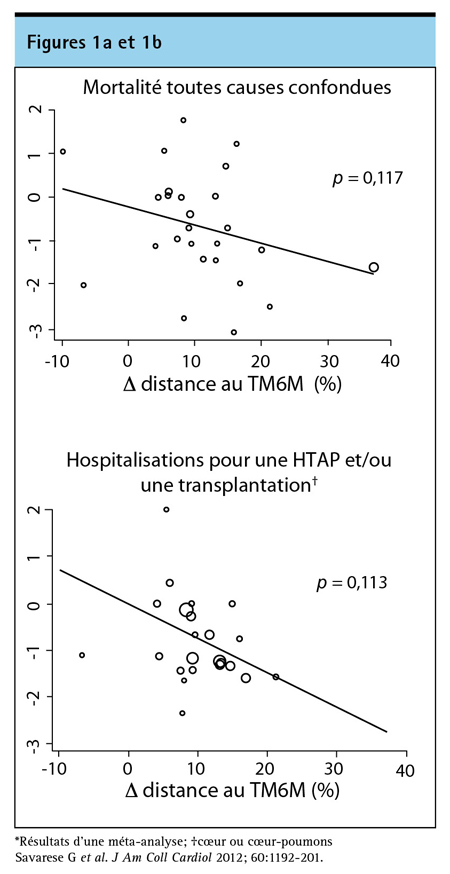
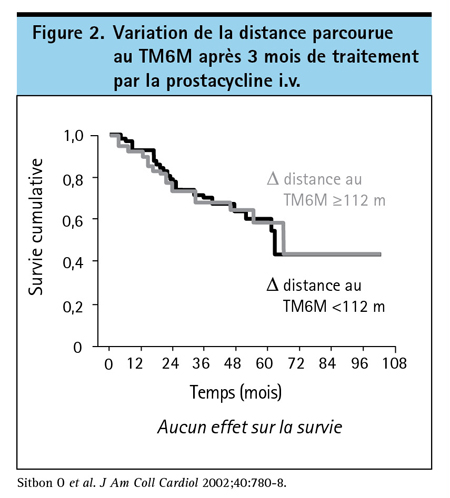
Ce n’est pas l’utilité de la distance parcourue au TM6M en tant que valeur initiale que l’on remet en question – cette dernière étant corrélée avec l’état fonctionnel et ayant peutêtre une valeur pronostique –, mais bien l’utilité de la variation de la distance parcourue au TM6M consécutive au traitement en tant que prédicteur de l’issue clinique. Dans un essai qui portait sur les facteurs pronostiques chez des patients sous époprosténol, une faible distance initiale parcourue au TM6M et d’autres preuves d’une maladie avancée étaient des facteurs pronostiques, alors que la variation de la distance parcourue au TM6M – contrairement à l’amélioration d’autres paramètres comme la classe fonctionnelle de la New York Heart Association (NYHA)16 – ne l’était pas. Dans cette étude, les courbes de mortalité étaient superposables lorsqu’on comparait une amélioration égale ou supérieure à 112 m et une amélioration inférieure à 112 m (Figure 2). Plus récemment, la distance initiale parcourue au TM6M a été prédictive de la mortalité à 2 ans dans deux essais avec randomisation sur l’ambrisentan, mais la variation de la distance parcourue au TM6M à 12 semaines ne l’était pas17.
Changement du contexte thérapeutique
Avant l’ère des traitements vasoactifs, l’HTAP était souvent détectée à un stade avancé, lorsque la capacité fonctionnelle était déjà fortement diminuée et que la probabilité de survie à 5 ans était faible18. Aujourd’hui, l’HTAP est plus souvent détectée à un stade assez précoce et, à en juger par une métaanalyse, les traitements sont associés à un gain de survie19. De nombreuses formes d’HTAP demeurent progressives et mortelles20, mais le nombre croissant de stratégies de traitement pouvant prolonger la survie donne tout lieu de croire que les essais cliniques devraient reposer sur un paramètre principal qui ferait ressortir une différence selon des paramètres majeurs, la survie en particulier.
Des experts convoqués pour discuter du plan et des paramètres des essais cliniques en sont venus à une conclusion similaire11. Bien qu’ils reconnaissent l’utilité de plusieurs paramètres conçus pour faire ressortir un changement au niveau des fonctions pulmonaire et vasculaire de même qu’une amélioration de la qualité de vie, les experts de ce groupe de travail recommandent que les essais pivots reposent sur un paramètre mixte permettant d’évaluer le délai d’aggravation clinique dont la définition serait uniforme. La définition proposée tient compte non seulement du résultat au TM6M défini au sens strict et corrélé avec la classe fonctionnelle, mais également de la mortalité toutes causes confondues et des hospitalisations liées à l’HTAP. Le groupe de travail a également recommandé que ces résultats soient évalués par un comité central.
Ces principes ont déjà été mis en application dans quelques essais cliniques, dont l’essai SERAPHIN sur le macitentan (antagoniste des récepteurs de l’endothéline) qui vient de se terminer21. Selon toutes probabilités, ces paramètres seront plus révélateurs de l’utilité relative des options commercialisées.
Résumé
Dans l’HTAP, le choix du traitement doit tenir compte de l’étiologie de la maladie, de la classe fonctionnelle du patient et des objectifs du traitement. Le TM6M demeure un outil simple et utile d’évaluation de la capacité fonctionnelle, mais il semble moins utile comme paramètre de substitution pour l’évaluation de l’effet du traitement sur la progression de la maladie lorsque deux stratégies actives sont comparées. Dans le cadre d’un essai clinique, des paramètres mixtes plus complets incluant des événements majeurs permettront sans doute de mieux capter les mérites relatifs des nouvelles options de traitement.
Références
1. Rubin LJ. The 6-minute walk test in pulmonary arterial hypertension: how far is enough? Am J Respir Crit Care Med 2012;186:396-7.
2. Solway S, Brooks D, Lacasse Y, Thomas S. A qualitative systematic overview of the measurement properties of functional walk tests used in the cardiorespiratory domain. Chest 2001;119:256-70.
3. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002;166:111-7.
4. Spruit MA, Polkey MI, Celli B et al. Predicting outcomes from 6-minute walk distance in chronic obstructive pulmonary disease. J Am Med Dir Assoc 2012;13:291-7.
5. Borel B, Provencher S, Saey D, Maltais F. Responsiveness of Various Exercise-Testing Protocols to Therapeutic Interventions in COPD. Pulmonary Medicine 2013;2013:410748.
6. Schoindre Y, Meune C, Dinh-Xuan AT, Avouac J, Kahan A, Allanore Y. Lack of specificity of the 6-minute walk test as an outcome measure for patients with systemic sclerosis. J Rheumatol 2009;36:1481-5.
7. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation 2006;114:1417-31.
8. Simonneau G, Galiè N, Rubin LJ et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43:5S-12S.
9. McLaughlin VV, Presberg KW, Doyle RL et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004;126:78S-92S.
10. Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001;104:429-35.
11. McLaughlin VV, Badesch DB, Delcroix M et al. End points and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54:S97-107.
12. Committee for Medicinal Products for Human Use: Guidelines on the clinical investigations of medicinal products for the treatment of pulmonary arterial hypertension. EMA, 2009. (Consulté le 29 août 2013; adresse : http://www.emea.europa.eu/ guideline/2009/12/WC500016686.pdf.).
13. Galiè N, Simonneau G, Barst RJ, Badesch D, Rubin L. Clinical worsening in trials of pulmonary arterial hypertension: results and implications. Curr Opin Pulm Med 2010;16 Suppl 1:S11-9.
14. Savarese G, Paolillo S, Costanzo P et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol 2012;60: 1192-201.
15. Gabler NB, French B, Strom BL et al. Validation of 6-minute walk distance as a surrogate end point in pulmonary arterial hypertension trials. Circulation 2012;126:349-56.
16. Sitbon O, Humbert M, Nunes H et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002;40:780-8.
17. Fritz JS, Blair C, Oudiz RJ et al. Baseline and followup 6-min walk distance and brain natriuretic peptide predict 2-year mortality in pulmonary arterial hypertension. Chest 2013;143:315-23.
18. D’Alonzo GE, Barst RJ, Ayres SM et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343-9.
19. Galiè N, Manes A, Negro L, Palazzini M, Bacchi- Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009;30:394-403.
20. Humbert M. Impression, sunset. Circulation 2013;127:1098-100.
21. Pulido T, Adzerikho I, Channick RN et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18.
Délai d’aggravation clinique : un paramètre à définir
David Langleben, MD, FRCPC
Directeur, Centre des maladies vasculaires pulmonaires
Hôpital général juif
Professeur titulaire de médecine
Université McGill
Montréal (Québec)
Le délai d’aggravation clinique (DAC) a été un paramètre secondaire commun aux essais de phase III ayant évalué l’efficacité des agents contemporains dans le traitement de l’hypertension artérielle pulmonaire (HTAP). Contrairement au test de marche de 6 minutes (TM6M), dont le résultat reflète l’évolution des symptômes, le DAC peut témoigner de l’effet du traitement sur un éventail beaucoup plus vaste de paramètres et, en particulier, sur la protection contre des événements, telles les hospitalisations ou la mort, qui caractérisent la progression de l’HTAP. Vu l’absence de traitements curatifs, il est tout à fait justifié d’évaluer les effets du traitement sur des paramètres majeurs, comme la mortalité ou les hospitalisations liées à l’HTAP, ainsi que sur la qualité de vie et la capacité fonctionnelle. En tant que paramètre principal, le DAC peut cerner les effets du traitement sur plusieurs paramètres pertinents, mais encore faut-il que les définitions des événements inclus dans le DAC soient uniformes et reproductibles. À mesure que les traitements se perfectionneront, il sera de plus en plus important de déterminer si ces derniers peuvent repousser les jalons de la progression de la maladie, surtout si l’on compare deux stratégies de traitement.
Objectifs actuels du traitement
L’HTAP appartient à l’une des cinq classes d’hypertension pulmonaire (HP), terme défini selon des critères hémodynamiques qui englobe un vaste éventail de physiopathologies1. Une HTAP peut se développer en présence de certaines maladies précipitantes, comme un shunt cardiaque congénital, une infection à VIH, une hypertension portale ou une maladie des tissus conjonctifs, et d’autres causes. L’incidence de l’HTAP idiopathique est de 1 ou 2 cas par million d’habitants2. Une prédisposition génétique pourrait jouer un rôle important dans l’HTAP, quoique l‘interaction entre certaines mutations et certains facteurs déclenchants environnementaux n’ait pas encore été totalement élucidée3. Il est possible, voire probable, que différentes étiologies aboutissant à l’HTAP donnent lieu à des processus physiopathologiques similaires qui entraînent un remodelage et une sténose des microvaisseaux pulmonaires.
Des différences dans l’activation ou la désactivation relative de multiples voies biochimiques impliquées dans le remodelage tissulaire associé à l’HTAP pourraient expliquer que la maladie n’évolue pas toujours à la même vitesse. Bien que tous les traitements commercialisés exercent des effets hémodynamiques similaires, les différentes voies qu’ils empruntent pour y arriver pourraient influer sur d’autres voies de signalisation moléculaire. Par exemple, l’époprosténol, tout comme d’autres prostacyclines ou analogues de la prostacycline, se fixe au récepteur de la prostacycline pour lancer une série d’étapes moléculaires qui, en activant les protéines G et en régulant l’AMPc à la hausse, aboutissent à la vasodilatation4. Ces agents exercent toutefois d’autres actions, en particulier des effets antiplaquettaires qui pourraient aussi contribuer à la maîtrise de l’HTAP, et peuvent modifier la production pulmonaire d’endothéline-15,6.
De même, les antagonistes des récepteurs de l’endothéline (ARE) bloquent les récepteurs siégeant sur les cellules endothéliales responsables de la libération d’endothéline, puissant vasoconstricteur et mitogène, mais ce système de récepteurs semble inter-relié avec des voies moléculaires indépendantes de la pression qui peuvent contribuer à la progression de l’HTAP7. Ces voies sont associées à l’activation pro-inflammatoire de cytokines et à des mécanismes favorisant la prolifération comme l’inhibition de l’apoptose8. Les effets relatifs des divers ARE pourraient différer selon que l’ARE cible le récepteur A de l’endothéline (ETA), qui semble participer principalement à la prolifération, ou le récepteur B (ETB), qui semble assurer principalement la vasodilatation et l’élimination de l’endothéline7. Les différences cliniques entre le blocage non sélectif et le blocage sélectif de ces récepteurs n’ont pas fait l’objet d’une étude approfondie9.
Les inhibiteurs de la phosphodiestérase de type 5 (PDE-5) exercent un effet vasodilatateur en bloquant l’hydrolyse de la guanosine monophosphate cyclique (GMPc), qui relâche les cellules musculaires lisses10. Le premier stimulateur de la guanylate cyclase soluble à être homologué, le riociguat, stimule directement la production de GMPc11. Le lien entre une activité accrue de la GMPc et des effets prolifératifs pourrait ici encore jouer un rôle dans l’inhibition du remodelage des artères pulmonaires12.
La plupart des agents actuellement utilisés dans le traitement de l’HTAP ont été homologués sur la foi de leur effet favorable sur la distance parcourue au TM6M par rapport à un placebo. Chacun de ces agents exerce vraisemblablement un effet sur de nombreuses voies intervenant dans la progression de la maladie. Comparativement à un paramètre majeur unique, la mortalité par exemple, qui nécessiterait la tenue d’essais d’envergure de très longue durée, un paramètre mixte comme le DAC – qui englobe à la fois la capacité fonctionnelle et la survie sans événement – a l’avantage d’être viable dans le cadre d’essais d’envergure et de durée moindres. Les deux paramètres inclus dans le DAC témoignent du bien-être des patients.
Le DAC en tant que paramètre Clinique
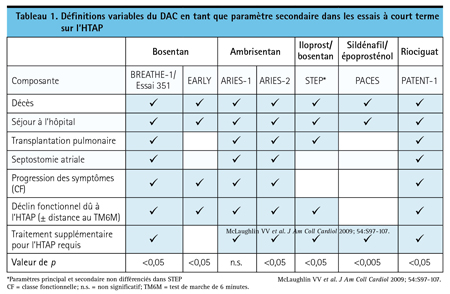
Le DAC figure parmi les paramètres secondaires des essais cliniques sur l’HTAP depuis une dizaine d’années. Dans BREATHE-1, essai comparatif avec placebo qui a mené à l’homologation du bosentan, le DAC était un paramètre mixte qui regroupait la mort, la transplantation pulmonaire, l’hospitalisation pour cause d’hypertension pulmonaire, l’absence d’amélioration clinique ou une aggravation entraînant l’abandon du traitement, la nécessité d’un traitement par l’époprosténol et une septostomie atriale13. Dans ARIES-1, essai comparatif avec placebo qui a mené à l’homologation de l’ambrisentan, la définition du DAC était similaire, mais les patients qui avaient besoin de médicaments supplémentaires en raison d’une aggravation clinique de leur HTAP étaient retirés de l’étude14. Le DAC a aussi été un paramètre secondaire d’autres essais comme EARLY (bosentan dans l’HTAP peu avancée), PACES (association sildénafil + époprosténol) et PATENT-1 (efficacité du riociguat), mais sa définition variait légèrement d’un essai à l’autre15-17 (Tableau 1).
Comparativement à un paramètre unique, qu’il s’agisse de la capacité d’exercice ou d’un événement particulier comme la mort, le DAC – du fait qu’il regroupe plusieurs éléments – permet une évaluation plus complète des retombées du traitement. Cependant, la combinaison de paramètres majeurs tels que l’hospitalisation ou la mort et de paramètres moins tangibles comme le degré d’aggravation justifiant l’ajout d’un traitement a le désavantage d’ouvrir la porte à des définitions et à une adjudication de rigueur variable. En outre, il peut être impossible de déterminer si le traitement est utile pour une seule des composantes du paramètre mixte, car la puissance statistique de l’essai repose sur l’analyse globale du paramètre mixte. Cela dit, au même titre qu’une analyse en intention de traiter prévue pour éviter de fausser les données après l’attribution du traitement, le DAC témoigne d’un effet global du traitement sur l’évolution clinique au sein d’une population donnée.
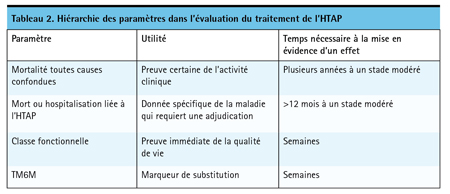
Le cadre temporel est important. Souvent, les essais sur l’efficacité d’un traitement dans l’HTAP évaluent l’amélioration de la capacité d’exercice sur une brève période, typiquement moins de 6 mois. Au vu du nombre croissant de traitements oraux efficaces que l’on peut instaurer à un stade plus précoce de la maladie, il devient plus intéressant d’évaluer l’effet du traitement sur la progression de la maladie même si la mise en évidence d’un effet nécessite un suivi plus long (Tableau 2). Les premiers essais sur les traitements à base d’une prostacycline administrés par voie intraveineuse (i.v.) ciblaient des patients dont l’HTAP était avancée et chez qui il était urgent et prioritaire de soulager les symptômes, mais nous avons maintenant la possibilité d’interrompre la physiopathologie de l’HTAP avant que l’atteinte structurale ne soit généralisée, ce qui, avec le temps, pourrait aboutir au développement de traitements qui maîtrisent le processus morbide sous-jacent.
Essais cliniques
Le DAC a été promu au rang de paramètre principal dans plusieurs essais en cours et dans un essai déjà terminé. Dans le premier essai mené à terme, SERAPHIN, le DAC se définissait par la survenue de l’un des événements suivants : mort, septostomie atriale, transplantation pulmonaire, instauration d’un traitement par un prostanoïde par voie i.v. ou sous-cutanée ou aggravation de l’HTAP18. Les patients – dont environ la moitié étaient de classe fonctionnelle (CF) II selon l’Organisation mondiale de la Santé (OMS) au début du traitement – ont été randomisés de façon à recevoir une ou deux doses de macitentan (ARE) ou un placebo. Au cours d’un suivi d’une durée moyenne d’environ 2 ans, la dose de 10 mg de macitentan a été associée à une réduction de 45 % (p<0,001) du risque d’aggravation clinique par rapport au placebo.
Dans cet essai, la plus forte des deux doses de macitentan a été associée à une réduction non significative de la mortalité liée à l’HTAP par rapport au placebo, mais l’effet du traitement tenait en grande partie à une incidence moindre d’événements dénotant une aggravation de l’HTAP. La mort était rarement le premier événement, ce qui n’a rien d’étonnant dans un essai où les cas d’HTAP légère à modérée sont majoritaires (Figure 1). Cet essai axé sur la survenue d’événements étant le premier à objectiver la supériorité d’un traitement par rapport à un placebo dans l’HTAP, on ignore si d’autres traitements donneraient d’aussi bons résultats. Compte tenu du nombre élevé de traitements maintenant commercialisés pour l’HTAP, nous avons besoin de telles données pour concevoir des algorithmes rationnels. COMPASS-2, un autre essai axé sur la survenue d’événements dont les patients ont été randomisés de façon à recevoir du sildénafil (inhibiteur de la PDE-5) seul ou en association avec du bosentan (ARE), nous aidera peut-être en ce sens19.
La standardisation des définitions du DAC nous sera utile pour comparer des stratégies de traitement, mais une comparaison d’agents devrait toujours se faire dans le cadre d’un essai et non d’un essai à l’autre. Un groupe de travail de l’OMS, qui a envisagé la possibilité d’une standardisation des définitions, a été le premier à se pencher sur la question en 200820. Les experts de ce groupe de travail ont recommandé en particulier que la mortalité, les hospitalisations non ajournables et le déclin de la capacité d’exercice rigoureusement confirmé fassent partie du DAC, mais ont concédé que d’autres aspects du bénéfice clinique, en particulier un changement dans la qualité de vie, étaient importants (Figure 1). Le groupe d’experts a en outre recommandé que l’adjudication des données soit confirmée par un comité central afin que les données soient encore plus solides, mais il a aussi reconnu que l’HTAP est une maladie complexe dont le traitement peut être d’une utilité variable selon que la maladie est avancée ou non.

L’HTAP se caractérise par une élévation de la pression artérielle pulmonaire, certes, mais ce sont les changements structuraux survenant dans les poumons et la sévérité de la dysfonction cardiaque qui déterminent le risque d’événement. L’importance relative des processus moléculaires interdépendants impliqués dans ces changements structuraux pourrait différer selon l’étiologie de l’HTAP ou le patient. Dans la mesure où les traitements à notre disposition pourraient exercer une influence variable sur ces processus, des agents qui améliorent la capacité d’exercice de façon similaire pourraient ne pas conférer la même protection contre la progression de la maladie exprimée par la survenue d’événements ou les processus sous-jacents comme le remodelage tissulaire. En effet, même le plus puissant des traitements pourrait ne pas influer sur le remodelage des microvaisseaux pulmonaires21. En pareil cas, le simple fait de retarder la survenue d’un événement clinique pourrait revêtir une importance critique.
Résumé
L’HTAP est une maladie progressive qu’il est impossible de faire régresser à l’aide des traitements actuels, mais des données montrent qu’il est possible de stabiliser l’évolution clinique pendant au moins plusieurs années chez de nombreux patients. Au nombre de ces données figurent les résultats favorables d’un essai de phase III dont le paramètre principal était le DAC. Comparativement à la capacité d’exercice mesurée par le TM6M, le DAC évalue la possibilité que les traitements actuels ralentissent ou stoppent la progression de l’HTAP et qu’ils atténuent les symptômes. En tant que paramètre d’un essai clinique, le DAC est d’autant plus utile qu’à défaut d’un traitement curatif, la stabilisation de l’état clinique ou l’amélioration des fonctions pulmonaire et cardiaque sont les objectifs ultimes du traitement. Dans les essais menés à ce jour, la définition du DAC varie selon que ce dernier est un paramètre principal ou secondaire, mais des définitions uniformes faciliteraient les grands efforts que l’on déploie pour hiérarchiser les interventions.
Références
1. Galiè N, Hoeper MM, Humbert M et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30: 2493-537.
2. Humbert M, Sitbon O, Chaouat A et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173:1023-30.
3. Yuan JX, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Circulation 2005;111:534-8.
4. Ruan CH, Dixon RA, Willerson JT, Ruan KH. Prostacyclin therapy for pulmonary arterial hypertension. Texas Heart Institute Journal / from the Texas Heart Institute of St Luke’s Episcopal Hospital, Texas Children’s Hospital 2010;37:391-9.
5. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106:1477-82.
6. Langleben D, Barst RJ, Badesch D et al. Continuous infusion of epoprostenol improves the net balance between pulmonary endothelin-1 clearance and release in primary pulmonary hypertension. Circulation 1999;99:3266-71.
7. Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 2000;102:2434-40.
8. Shichiri M, Kato H, Marumo F, Hirata Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension 1997;30:1198-203.
9. Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PloS one 2012;7:e47662.
10. Wilkins MR, Wharton J, Grimminger F, Ghofrani HA. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur Resp J 2008;32:198-209.
11. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 2011;123: 2263-73.
12.Wharton J, Strange JW, Moller GM et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med 2005;172:105-13.
13. Rubin LJ, Badesch DB, Barst RJ et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903.
14. Galiè N, Olschewski H, Oudiz RJ et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebocontrolled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010-9.
15. Galiè N, Rubin L, Hoeper M et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008;371:2093-100.
16. Simonneau G, Rubin LJ, Galiè N et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521-30.
17. Ghofrani HA, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330-40.
18. Pulido T, Adzerikho I, Channick RN et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18.
19. clinicaltrials.gov. Effects of the combination of bosentan and sildenafil versus sildenafil monotherapy on pulmonary afterial hypertension (PAH) (COMPASS-2). http://clinicaltrialsgov/show/ NCT003034592006.
20. McLaughlin VV, Badesch DB, Delcroix M et al. End points and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54:S97-107.
21. Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg- Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 2010;138:1234-9.
Monothérapie vs traitement d’association : efficacité selon les paramètres d’évaluation de l’HTAP
Robert D. Levy, MD, FRCPC
Codirecteur, Programme d’hypertension pulmonaire
Vancouver General Hospital
Professeur titulaire de médecine, Division de pneumologie
University of British Columbia
Vancouver (Colombie-Britannique)
Dans l’hypertension artérielle pulmonaire (HTAP), l’association de deux agents efficaces dotés de modes d’action distincts devrait logiquement être plus bénéfique qu’une monothérapie. Selon plusieurs guides de pratique publiés, il est pertinent d’envisager un traitement d’association si la réponse à un agent unique est insuffisante, mais on précise qu’aucune preuve n’étaye cette pratique. L’absence de preuves tient non seulement à un manque d’essais cliniques, mais aussi aux résultats contradictoires d’études de faible envergure. Plusieurs facteurs pourraient expliquer le manque de constance dans les résultats : efficacité inégale de différentes associations, hétérogénéité des populations de patients, sévérité variable de la maladie au moment de l’administration du traitement d’association et différences dans l’évaluation du bénéfice clinique. À défaut d’un traitement curatif, le meilleur moyen de retarder la progression réside peut-être en l’association d’agents exerçant une action sur différents mécanismes physiopathologiques de l’HTAP, mais cette hypothèse devra être vérifiée.
Objectifs actuels du traitement
À l’heure actuelle, le traitement de l’hypertension pulmonaire repose sur les classes suivantes de médicaments : prostanoïdes, antagonistes des récepteurs de l’endothéline (ARE), inhibiteurs de la phosphodiestérase de type 5 (PDE-5), inhibiteurs calciques et, depuis peu, stimulateurs de la guanylate cyclase soluble (GCs). Jusqu’à tout récemment, l’efficacité de ces agents était le plus souvent évaluée à l’aide du test de marche de 6 minutes (TM6M)1. La plupart des essais ayant porté sur des associations ont aussi utilisé ce paramètre principal2. L’HTAP étant une maladie évolutive qui se caractérise par des lésions cardiaques et vasculaires structurelles menant à une détérioration irréversible de la fonction cardiopulmonaire3, il est maintenant recommandé que tous les essais visant à évaluer l’efficacité de médicaments reposent sur des paramètres cliniques majeurs, comme l’hospitalisation et la mortalité liées à l’HTAP4, à plus forte raison pour mettre en évidence un avantage du traitement d’association. Dans les cas d’HTAP avancée, où le traitement d’association pourrait logiquement être le plus intéressant, l’amélioration de la capacité d’exercice n’est peut-être pas un marqueur sensible d’un bénéfice cliniquement significatif.
Bien que la biopathologie de l’HTAP n’ait pas été totalement élucidée, tous les agents à notre disposition ciblent des médiateurs vasoactifs afin de favoriser une vasodilatation (et possiblement une inhibition de la prolifération), bloquant ainsi le remodelage structurel des vaisseaux pulmonaires5. Chaque classe d’agents emprunte toutefois une voie différente pour y arriver (Figure 1). Les prostanoïdes sont de puissants vasodilatateurs que l’on administre pour suppléer à la prostacycline endogène, dont la production fait défaut chez les patients atteints d’HTAP6. Les ARE bloquent la fixation de l’endothéline-1, puissant vasoconstricteur régulé à la hausse dans l’HTAP7. Les inhibiteurs de la PDE-5 bloquent l’activité d’une enzyme qui inhibe la guanosine monophosphate cyclique (GMPc), médiateur clé du monoxyde d’azote (NO) connu pour ses propriétés vasodilatatrices8. Les stimulateurs de la GCs ont aussi pour effet de faire augmenter le taux de GMPc afin de favoriser la vasorelaxation, mais ils y parviennent au moyen d’un mécanisme indépendant de l’activité du NO9. Dans tous les cas, la diminution de la vasoconstriction pulmonaire a été liée aux effets antiprolifératifs essentiels au ralentissement ou à la prévention du remodelage tissulaire qui caractérise la progression de la maladie.
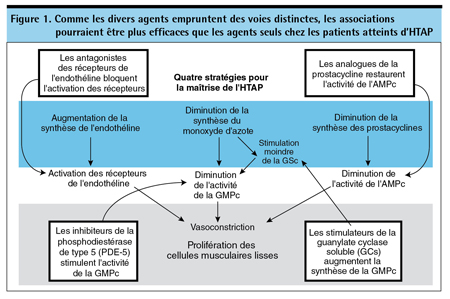
Les données montrant que les agents actuels peuvent ralentir la progression de la maladie lorsqu’ils sont employés seuls proviennent de plusieurs sources, dont une méta-analyse d’essais comparatifs ayant objectivé un gain de survie10. En outre, lors d’essais cliniques comparatifs, on a souvent observé des effets favorables du traitement sur des paramètres secondaires ayant une valeur pronostique, notamment une augmentation de l’index cardiaque, une diminution du taux sérique de peptide B-natriurétique (BNP) et une diminution durable des résistances vasculaires pulmonaires (RVP). Récemment, le premier essai de phase III dans l’HTAP à utiliser des événements cliniques comme paramètre principal a également généré des résultats favorables. Lors de cet essai, intitulé SERAPHIN, un nouvel ARE ciblant les deux types de récepteurs de l’endothéline – le macitentan – a été associé, comparativement à un placebo, à une diminution de 45 % des événements compris dans le paramètre mixte, à savoir : décès, septostomie atriale, transplantation pulmonaire, instauration d’un traitement par un prostanoïde intraveineux ou sous-cutané ou aggravation de l’HTAP après une période d’une durée moyenne de 2 ans11. En tant que paramètre isolé, la mortalité ne différait pas significativement.
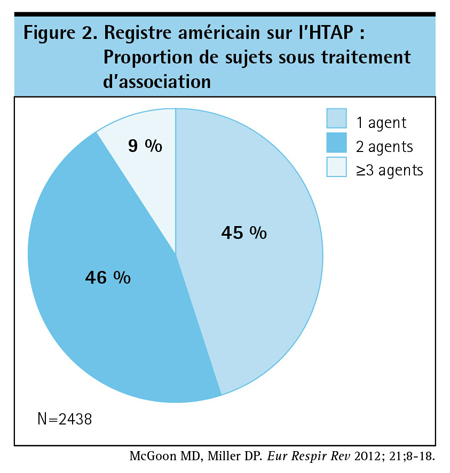
Si les agents de chacune des classes utilisées dans le traitement de l’HTAP emprunte une voie distincte pour ralentir la progression de la maladie, il est raisonnable de s’attendre à ce qu’une association d’agents ait des effets additifs, voire synergiques. Cette théorie a beaucoup d’adeptes malgré le peu de données l’étayant. Un sondage mené auprès de 2438 patients inscrits dans le plus vaste registre américain sur l’HTAP (réunissant uniquement des sujets recevant un médicament pour l’HTAP) a révélé que 46 % et 9 % des répondants recevaient respectivement deux et trois agents12 (Figure 2). Dans plusieurs guides de pratique, on aborde la possibilité d’un avantage du traitement d’association en l’absence d’une réponse suffisante à une monothérapie tout en reconnaissant l’insuffisance des preuves13,14.
Essais cliniques sur le traitement d’association
Les essais où l’on a évalué un traitement d’association avaient une population variant entre 33 et 405 patients, portaient sur différentes associations dont la séquence était variable et étaient de courte durée pour la plupart (Tableau 1). Bien que de nombreux essais aient été réalisés chez des patients dont la maladie était avancée – de classe fonctionnelle (CF) III ou IV selon l’Organisation mondiale de la Santé (OMS) –, l’essai EARLY a objectivé une amélioration de la capacité d’exercice chez des patients de CF II (OMS) qui prenaient déjà du sildénafil lorsqu’ils ont reçu du bosentan plutôt qu’un placebo15. Le TM6M était le paramètre principal d’évaluation de l’efficacité dans la plupart de ces essais, mais beaucoup d’entre eux accordaient autant d’importance à l’innocuité, en particulier aux interactions pharmacocinétiques.
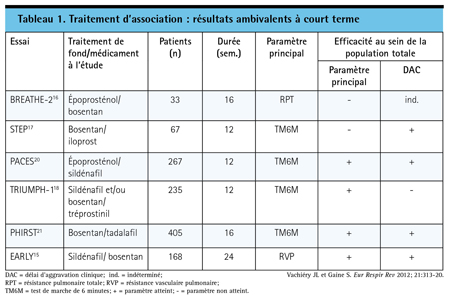
Comme les résultats du traitement d’association de quelques séries de cas semblaient prometteurs, BREATHE-2 – le premier essai comparatif mené à double insu avec placebo – a été d’envergure modeste16. Lors de cet essai, 33 patients atteints d’HTAP de CF III ou IV selon l’OMS ont d’abord reçu de l’époprosténol (prostanoïde), puis ont été randomisés à 16 semaines de façon à recevoir en plus de leur traitement soit du bosentan (ARE), soit un placebo. Après 16 semaines, les chercheurs ont noté une amélioration des paramètres hémodynamiques et de la capacité d’exercice, mais l’écart entre les deux groupes n’a pas atteint le seuil de significativité statistique.
BREATHE-2, dont les résultats sont considérés comme non concluants plutôt que défavorables, a pavé la voie à une série d’essais, notamment STEP17, COMBI18, et TRIUMPH19, qui incluaient tous un prostanoïde. Les résultats sont demeurés ambivalents. Dans le cadre de l’essai STEP, 67 patients atteints d’HTAP de CF III selon l’OMS ont d’abord reçu du bosentan avant d’être randomisés de façon à recevoir de l’iloprost en inhalation ou un placebo. À 12 semaines, la distance parcourue au TM6M était significativement plus longue chez les patients randomisés de façon à recevoir de l’iloprost en plus du bosentan que chez les patients randomisés de façon à recevoir un placebo en plus du bostentan17. La probabilité d’amélioration de la CF était significativement plus élevée chez les patients recevant la bithérapie active; de plus, les chercheurs ont observé une amélioration significative – par rapport au placebo – de quelques paramètres hémodynamiques comme la pression artérielle pulmonaire moyenne (PAPm) et la RVP. L’essai COMBI18, qui évaluait la même association, a pris fin lorsqu’une analyse intermédiaire a révélé que l’ajout de l’iloprost en inhalation par rapport au bosentan seul n’avait aucunement amélioré les résultats au TM6M (paramètre principal) chez les 40 patients inscrits jusque-là.
Le paramètre principal (distance parcourue au TM6M) a été atteint dans l’essai TRIUMPH, beaucoup plus vaste. Cependant, l’absence d’avantage significatif selon plusieurs paramètres secondaires importants aux yeux des patients – notamment l’amélioration de la CF, le délai d’aggravation clinique ainsi que la diminution des signes et symptômes de l’HTAP – complique l’interprétation des résultats. Dans cet essai, 235 patients atteints d’HTAP de CF III ou IV selon l’OMS qui recevaient soit du bosentan, soit du sildénafil (inhibiteur de la PDE-5), ont été randomisés de façon à recevoir du tréprostinil ou un placebo et ont été évalués après 12 semaines.
Dans un essai encore plus vaste, PACES, 267 patients atteints d’HTAP avancée qui recevaient une dose stable d’époprosténol ont été randomisés de façon à recevoir du sildénafil (inhibiteur de la PDE-5) ou un placebo20. Comparativement au placebo, l’association a amélioré significativement le résultat au TM6M, le paramètre principal, et a exercé un effet favorable sur plusieurs paramètres secondaires cliniquement importants, notamment le délai d’aggravation clinique et la qualité de vie liée à la santé. Le traitement a eu également un effet favorable sur plusieurs paramètres hémodynamiques, mais les auteurs ont noté que le bénéfice relatif était d’autant plus marqué que la capacité d’exercice était bonne au départ.
Dans le plus vaste des essais sur un traitement d’association, PHIRST, 405 patients atteints d’HTAP qui n’étaient pas déjà sous bosentan ont d’abord reçu du bosentan avant d’être randomisés de façon à recevoir l’une des doses évaluées de tadalafil (inhibiteur de la PDE-5) ou un placebo21. Comparativement au placebo, le tadalafil a amélioré la distance parcourue au TM6M, le paramètre principal, de manière significative et proportionnelle à la dose. En outre, toujours comparativement au placebo, l’association a retardé l’aggravation clinique et amélioré la qualité de vie liée à la santé. La majorité des patients de cet essai – comme ceux des essais antérieurs – étaient de CF III ou IV selon l’OMS, mais environ le tiers des patients étaient de CF II.
L’essai ouvert COMPASS-122, qui a objectivé des effets hémodynamiques favorables de l’association bosentan + sildénafil, a été suivi de l’essai COMPASS-2, qui tire maintenant à sa fin. Dans le cadre de cet essai mené à double insu avec placebo, 330 patients atteints d’HTAP ont été randomisés de façon à recevoir du bosentan seul, du sildénafil seul ou les deux en association. Le paramètre principal regroupe plusieurs événements cliniques.
Dans l’essai IMPRES qui vient de se terminer, l’imatinib, inhibiteur de la voie de signalisation du PDGF (platelet-derived growth factor), a été évalué chez des patients atteints d’HTAP qui recevaient déjà deux autres agents pour le traitement de l’HTAP23. Malgré un taux élevé d’effets indésirables graves ayant entraîné l’abandon du traitement chez 44 % des patients, les paramètres hémodynamiques et la capacité d’exercice s’étaient améliorés par rapport au placebo après 24 semaines. Il n’y avait aucune différence quant à la classe fonctionnelle, au délai d’aggravation clinique ou à la mortalité. Après que les autorités aient exigé plus de données sur l’efficacité et l’innocuité de l’imatinib dans l’HTAP, le promoteur a récemment décidé de retirer sa demande d’homologation dans cette indication, tant aux États-Unis que dans les pays de l’Union européenne.
Traitement d’association : la situation actuelle
Les traitements d’association demeurent prometteurs, bien qu’ils n’aient pas été totalement validés dans le contexte des algorithmes de traitement visant à améliorer les paramètres cliniques pertinents. Une métaanalyse a révélé que le traitement d’association donnait lieu à une légère amélioration de la capacité d’exercice comparativement à des agents en monothérapie, mais n’a pas apporté de preuves irréfutables d’une amélioration des résultats24. Cependant, les avantages théoriques de l’intensification du traitement au moyen d’une association demeurent incontestables. Nous avons besoin d’essais multicentriques axés sur la survenue d’événements au sein de populations bien définies aux stades précoce et avancé de la maladie. Certes, nous avons surtout besoin de traitements plus efficaces aux stades avancés de la maladie, les traitements traditionnels étant d’efficacité moindre en pareils cas, mais il est tout aussi important d’explorer l’hypothèse voulant qu’un traitement d’association précoce ralentisse la progression de la maladie. Nous avons besoin d’études pour déterminer si une maîtrise serrée plus précoce au moyen d’un traitement d’association en première intention est préférable à une intensification séquentielle du traitement lorsque les objectifs thérapeutiques ne sont pas atteints, mais on préconise d’atteindre les objectifs en surveillant le patient de près et en intensifiant le traitement lorsque les objectifs ne sont pas atteints25.
La décision d’avoir recours à un traitement d’association en l’absence de telles données est complexe et nécessite une évaluation minutieuse du rapport bénéfice:risque escompté. Dans les guides de pratique, le recours aux associations est accepté lorsque la réponse à la monothérapie est en perte de vitesse, mais on ne formule aucune recommandation précise quant à la nature de l’association à utiliser ni quant aux modalités d’utilisation. La possibilité d’atténuation des symptômes doit être soupesée en regard de la possibilité d’exacerbation des effets indésirables, mais les maigres données comparatives font obstacle à une solide démarche objective.
Résumé
L’HTAP est une maladie incurable qui évolue vers l’insuffisance cardiaque droite et la mort. Le premier essai clinique à être axé sur la survenue d’événements corrobore d’autres données montrant que les agents actuellement à notre disposition ralentissent la progression de la maladie, mais la morbidité et la mortalité demeurent élevées. La possibilité d’améliorer les résultats en combinant au moins deux agents actifs dotés de modes d’action distincts est bien sûr séduisante, mais nous manquons de preuves provenant d’essais comparatifs avec randomisation. Nous avons besoin d’essais axés sur les événements non seulement pour valider la pertinence du traitement d’association, mais aussi pour mieux comprendre comment en optimiser l’utilisation pour infléchir l’évolution naturelle de la maladie.
Références
1. Rich S. The 6-minute walk test as a primary endpoint in clinical trials for pulmonary hypertension. J Am Coll Cardiol 2012;60:1202-3.
2. Galiè N, Negro L, Simonneau G. The use of combination therapy in pulmonary arterial hypertension: new developments. European Respiratory Review: An official Journal of the European Respiratory Society 2009;18: 148-53.
3. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004;351:1655-65.
4. McLaughlin VV, Badesch DB, Delcroix M et al. End points and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54:S97-107.
5. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation 2004;109:159-65.
6. Galiè N, Manes A, Branzi A. Prostanoids for pulmonary arterial hypertension. American Journal of Respiratory Medicine: Drugs, Devices and Other Interventions 2003;2:123-37.
7. Galiè N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovascular Research 2004;61:227-37.
8. Galiè N, Ghofrani HA, Torbicki A et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148-57.
9. Schermuly RT, Janssen W, Weissmann N, Stasch JP, Grimminger F, Ghofrani HA. Riociguat for the treatment of pulmonary hypertension. Expert Opinion on Investigational Drugs 2011;20:567-76.
10. Galiè N, Manes A, Negro L, Palazzini M, Bacchi- Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009;30:394-403.
11. Pulido T, Adzerikho I, Channick RN et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18.
12. McGoon MD, Miller DP. REVEAL: a contemporary US pulmonary arterial hypertension registry. European Respiratory Review: An official Journal of the European Respiratory Society 2012;21:8-18.
13. McLaughlin VV, Archer SL, Badesch DB et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009;119: 2250-94.
14. Galiè N, Hoeper MM, Humbert M et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493-537.
15. Galiè N, Rubin LJ, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomized controlled trial. Lancet 2008;371:2093-2100.
16. Humbert M, Barst RJ, Robbins IM et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Resp J 2004;24:353-9.
17. McLaughlin VV, Oudiz RJ, Frost A et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:1257-63.
18. Hoeper MM, Leuchte H, Halank M et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur Resp J 2006;28:691-4.
19. McLaughlin VV, Benza RL, Rubin LJ et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010;55:1915-22.
20. Simonneau G, Rubin LJ, Galiè N et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521-30.
21. Galiè N, Brundage BH, Ghofrani HA et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119:2894-903.
22. Greunig M, Michelakis A, Vachiery JL. Eur Heart J 2007;28:140.
23. Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary hypertension: results of the randomized IMPRES study. Circulation 2013;127:1128-38.
24. Fox BD, Shimony A, Langleben D. Meta-analysis of monotherapy versus combination therapy for pulmonary arterial hypertension. American Journal of Cardiology 2011;108:1177-82.
25. Hoeper MM. “Treat-to-target” in pulmonary arterial hypertension and the use of extracorporeal membrane oxygenation as a bridge to transplantation. Eur Respir Rev 2011;20:297-300.
Survie et qualité de vie : objectifs pertinents à redéfinir dans l’HTAP
Sanjay Mehta, MD, FRCPC, FCCP
Président, Conseil d’administration, Association d’hypertension pulmonaire du Canada
Directeur, Clinique d’hypertension pulmonaire du sud-ouest de l’Ontario
Professeur titulaire de médecine
Division de pneumologie / Département de médecine
Schulich School of Medicine & Dentistry
Western University
London (Ontario)
Grave et généralement évolutive, l’hypertension artérielle pulmonaire (HTAP) est souvent mortelle. Avant l’avènement d’agents homologués expressément pour le traitement de l’HTAP, la vie des patients était extrêmement limitée; ces derniers étaient souvent handicapés par des symptômes tels que la dyspnée et étaient exposés à un risque très élevé de décès quelques années après le diagnostic. Par exemple, la médiane de survie des patients atteints d’une HTAP primitive était de 2,8 ans à partir du diagnostic1, et moins du tiers des patients étaient toujours en vie après 5 ans. En cette ère moderne de la prise en charge de l’HTAP, nous estimons que la survie s’est améliorée légèrement2,3, ce qui a des répercussions sur les objectifs du traitement. Bien que l’HTAP demeure un processus morbide progressif et incurable, la capacité relative des stratégies thérapeutiques à atténuer cette progression mérite d’être évaluée indépendamment de leur capacité relative à maîtriser les symptômes. La survie prolongée des patients nous donne aussi la possibilité de comparer les retombées des diverses stratégies de traitement sur leur qualité de vie plus longtemps. Les essais comparatifs visant à comparer les agents de nouvelle génération avec un placebo comportent généralement un vaste éventail de paramètres secondaires exploratoires dont l’objectif est d’évaluer les effets du traitement. Comme il est maintenant possible de choisir parmi plusieurs stratégies pour le traitement de l’HTAP, l’effet relatif sur la survie et la morbidité revêt une importance grandissante.
Objectifs actuels du traitement
Bien que l’hypertension artérielle pulmonaire (HTAP) ait de multiples étiologies – malformation cardiaque congénitale, maladie des tissus conjonctifs et infection par le virus de l’immunodéficience humaine (VIH), notamment –, elle est idiopathique chez près de 50 % des patients des essais cliniques et dans la pratique clinique3. Cependant, quelle qu’en soit l’étiologie, sa physiopathologie est complexe : l’obstruction des vaisseaux pulmonaires résulte d’une vasoconstriction, de l’hypertrophie et la prolifération des cellules musculaires lisses, de la fibrose de l’intimamédia et de la thrombose microvasculaire, qui font toutes obstacle au débit sanguin dans les vaisseaux pulmonaires; la surcharge ventriculaire droite (VD) qui en résulte aboutit à une insuffisance VD et à la mort. Tous les agents homologués pour le traitement de l’HTAP se sont révélés capables d’atténuer les symptômes, vraisemblablement en diminuant la pression artérielle pulmonaire (PAP) et la résistance vasculaire pulmonaire (RVP) et, par conséquent, en améliorant la fonction VD et le débit sanguin dans les vaisseaux pulmonaires4 (Tableau 1).
Selon la classification de l’Organisation mondiale de la Santé (OMS) fondée sur le tableau clinique ou l’étiologie présumée, l’HTAP est l’un des cinq grands sous-types d’hypertension pulmonaire. Tous les soustypes se caractérisent par une élévation de la PAP qui altère la structure des poumons et du coeur. Pour déterminer les répercussions des symptômes découlant de ces altérations structurelles sur les activités de la vie quotidienne (AVQ), on a recours aux classes fonctionnelles (CF) modifiées de la New York Heart Association (NYHA) (I à IV).
Dans l’essai qui a mené à l’homologation de l’époprosténol, prostanoïde et premier agent conçu expressément pour le traitement de l’HTAP, l’objectif était de démontrer une amélioration fonctionnelle mesurée à l’aide du test de marche de 6 minutes (TM6M) sur une période de 12 semaines5. Jusqu’à tout récemment, la plupart des essais étaient axés sur le même paramètre principal que l’on mesurait sur une période de durée similaire (12 à 16 semaines), mais le nombre croissant d’options de traitement a amené les experts à choisir le traitement en fonction de paramètres cliniquement pertinents plus larges, dont la progression de la maladie. Au nombre de ces options figurent aujourd’hui, outre les prostanoïdes, des inhibiteurs calciques, des antagonistes des récepteurs de l’endothéline (ARE), des inhibiteurs de la phosphodiestérase de type 5 (PDE-5) et, depuis peu, un stimulateur de la guanylate cyclase soluble (GCs). Ces médicaments pour l’HTAP ont en commun le fait qu’ils abaissent la RVP, mais chacun y parvient en empruntant une voie distincte.
Compte tenu des altérations structurelles irréversibles caractéristiques de l’HTAP avancée, il est logique de viser un objectif différent à chacun des stades de la maladie. Lors des premiers essais, dont les sujets souffraient pour la plupart d’une HTAP avancée et terminale, il était peut-être raisonnable de viser un paramètre comme l’amélioration de la capacité fonctionnelle. Dans les stades moins avancés de la maladie, la capacité fonctionnelle n’est peut-être pas à elle seule représentative de la capacité du traitement à ralentir ou à stopper le processus morbide. Une incidence moindre d’événements cliniques sur une période de durée suffisante reflète mieux une diminution de la progression, mais des paramètres moléculaires, hémodynamiques ou structurels pourraient nous aider à déterminer si le traitement exerce un effet sur la physiopathologie sous-jacente et, le cas échéant, comment il y parvient.
Essais cliniques en HTAP : détermination du benefice
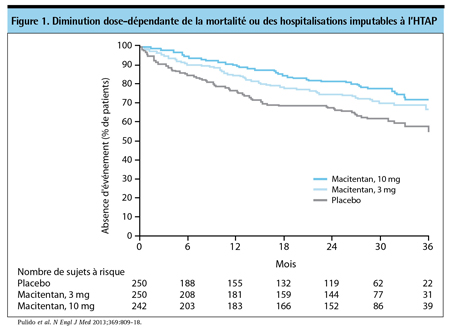
Il y a plusieurs années déjà, des experts ont recommandé que les essais cliniques permettent de mieux évaluer la maîtrise des symptômes et l’amélioration de la qualité de vie6. Le macitentan – un ARE innovateur – a fait l’objet du premier essai de phase III à utiliser un paramètre mixte englobant la morbidité et la mortalité pour évaluer l’effet du traitement, et les résultats de cet essai ont été publiés récemment7. Certes, 48,2 % des sujets étaient de CF III ou IV selon l’OMS, mais il reste qu’un peu plus de la moitié des sujets étaient de CF II, c’est-à-dire des sujets dont la capacité fonctionnelle était peu diminuée. La plus forte des deux doses de macitentan a diminué le risque relatif (HR) du risque d’aggravation clinique de 45 % (p<0,001). La diminution de 50 % (p<0,001) du risque de décès pour cause d’HTAP ou d’hospitalisation liée à l’HTAP – le paramètre secondaire mixte – par rapport au placebo signifie aussi que le traitement a ralenti la progression de la maladie, même si l’on ignore quel serait le bénéfice relatif du macitentan par comparaison à un autre agent actif (Figure 1).
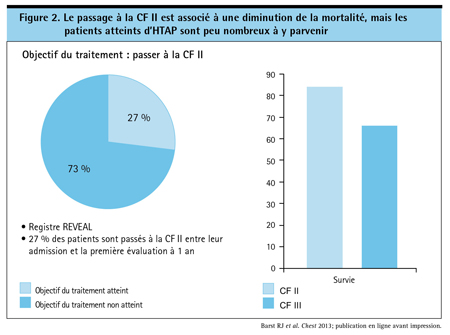
En raison des altérations structurelles progressives et irréversibles qui caractérisent l’HTAP, l’éventuel bénéfice clinique qui découle du traitement pourrait varier selon que la maladie est avancée ou non. Plus précisément, la probabilité de modification de l’évolution de la maladie pourrait être optimale avant la survenue d’une atteinte structurelle importante. Le ralentissement du processus morbide sous l’effet d’un traitement efficace, possible chez les patients dont la fonction pulmonaire et cardiaque est préservée, n’est peut-être plus possible en présence d’une atteinte fonctionnelle. Les données d’un registre ont permis de constater qu’une amélioration de la CF, marqueur potentiel de la réversibilité de la maladie, était prédictive de la survie à 3 ans8. Bien que la CF se soit améliorée chez seulement 27 % des sujets du registre, le taux de survie à 3 ans est passé de 66 % à 84 % (p<0,001) chez ceux qui sont passés de la CF III en début de traitement à la CF II (Figure 2).
Il est bien sûr impossible de comparer des stratégies de traitement sans faire d’essais cliniques, mais la complexité de l’HTAP est telle que des paramètres d’appoint pourraient être moins utiles pour déterminer l’effet de différentes options de traitement sur la progression de la maladie. Les améliorations à court terme de la CF de l’OMS et de la distance parcourue au TM6M – qui servent à évaluer l’efficacité des agents contemporains ne sont aucunement révélatrices des retombées du traitement de l’HTAP sur l’issue à long terme, telle l’hospitalisation pour une cause liée à l’HTAP. De plus, de tels paramètres à court terme sont de piètres paramètres de substitution quand il s’agit de déterminer si le traitement peut prévenir ou repousser la survenue de l’atteinte structurelle propre à l’HTAP. De même, l’amélioration d’autres paramètres de substitution, tels des biomarqueurs, ou la variation de paramètres biologiques pertinents dans l’HTAP, tels les critères hémodynamiques ou la protection contre une atteinte structurelle évolutive, ne nous sont pas très utiles pour juger de la supériorité clinique d’un agent sur un autre, bien que de tels paramètres puissent nous aider à comprendre comment différents agents ou différentes associations influent sur la physiopathologie de l’HTAP.
Dans l’HTAP, ce sont les objectifs du traitement qui déterminent en grande partie la méthodologie d’un essai clinique. Au départ, la priorité était de repérer des traitements capables de maîtriser les symptômes et, par conséquent, d’améliorer la capacité fonctionnelle. Depuis peu, on se tourne vers des essais cliniques conçus pour mettre en évidence une incidence moindre d’événements cliniquement pertinents à l’aide de paramètres mixtes révélateurs de la morbidité et de la mortalité, comme les hospitalisations liées à l’HTAP. Le jour où un essai clinique aura pour objectif de comparer deux stratégies de traitement actif plutôt qu’un agent actif et un placebo, les paramètres majeurs demeureront importants, mais des paramètres de substitution comme la protection relative contre le remodelage tissulaire nous aideraient à voir comment optimiser l’utilisation d’un traitement de façon à prévenir la progression de la maladie, surtout aux premiers stades de la maladie.
Optimisation du traitement
Dans les guides de pratique comme celui de la European Respiratory Society (ERS), la CF est un critère de sélection pour les traitements de première intention de l’HTAP9. En raison du nombre limité d’essais comparatifs, ces recommandations tiennent davantage à la méthodologie des essais qu’aux preuves d’une efficacité supérieure au sein de ces populations. Par exemple, en présence d’une importante vasoréactivité aiguë durant les évaluations hémodynamiques directes, on préconise un inhibiteur calcique de préférence à un agent d’une autre classe; par contre, en l’absence d’une vasoréactivité aiguë ou d’une réponse inappropriée à un inhibiteur calcique, on précise dans les guides de pratique qu’il n’y a aucune donnée concluante à l’appui d’une autre option de traitement.
Toujours dans les guides de pratique, on recommande d’évaluer régulièrement la progression de la maladie chez les patients sous traitement, mais les algorithmes de traitement donnent peu d’indications sur l’impact que la progression de la maladie devrait avoir sur le choix de traitement. Des tests cliniques, tel le TM6M, sont recommandés tous les 3 à 6 mois, alors que des examens plus sensibles pour détecter la progression de l’HTAP, comme l’échocardiographie et le cathétérisme cardiaque droit, sont recommandés dans l’éventualité d’une aggravation clinique ou dans les 6 mois suivant un changement de traitement. Le traitement d’association reposant sur un ARE, un inhibiteur de la PDE-5 ou un prostanoïde est recommandé dans l’éventualité d’une réponse insuffisante à l’un de ces agents en monothérapie, mais aucune donnée provenant d’un essai comparatif et prospectif n’étaye une association en particulier ou la supériorité d’une association par rapport à une autre.
Pour des raisons pratiques, les agents oraux sont préférables aux prostanoïdes, ces derniers devant être perfusés ou injectés. On le reconnaît dans de nombreux guides de pratique, dont celui de l’ERS, la plus haute cote (1-A) étant attribuée au bosentan, à l’ambrisentan et au sildénafil dans le traitement de l’HTAP chez des patients de CF II selon l’OMS. Dans le cas des patients de CF III, l’iloprost en inhalation et l’époprosténol injectable par voie intraveineuse (i.v.) s’ajoutent à ces agents. Par contre, seul l’époprosténol i.v. est étayé par des preuves 1-A pour le traitement de l’HTAP chez des patients de CF IV. Le tadalafil est le seul agent à avoir reçu une cote 1-B pour les patients de CF II et III; tous les autres agents oraux, seuls ou en association, sont étayés par des données IIa-C pour les patients de CF IV, car ils ont été peu évalués et se sont révélés peu avantageux dans la pratique clinique.
Cela dit, l’ERS et d’autres sociétés savantes insistent, à juste titre, que la prise en charge de l’HTAP ne peut pas se résumer à la prescription de traitements qui réduisent la pression vasculaire pulmonaire. Les soins de soutien, comme le soutien psychologique, la prévention des infections ainsi que l’usage de diurétiques et d’anticoagulants oraux, contribuent très étroitement à l’optimisation de la qualité de vie à mesure que la maladie s’aggrave. Une équipe multidisciplinaire peut être utile pour la mise sur pied d’un plan de traitement complet qui tient compte des complications éventuelles et des obstacles à l’accomplissement des activités de la vie quotidienne. Par exemple, les exercices de rééducation – qui sont prévus pour améliorer la capacité fonctionnelle même s’il n’a pas été démontré qu’ils amélioraient l’issue clinique – doivent être adaptés aux capacités du patient.
L’expansion des options de traitement et les efforts que l’on déploie pour détecter l’HTAP le plus tôt possible nous amènent à repenser aux objectifs du traitement. La possibilité d’un traitement curatif ne point pas à l’horizon, mais des études conçues pour comparer diverses stratégies quant à leur retombées sur les événements cliniques plutôt que sur la capacité fonctionnelle pourraient nous amener à déterminer quels traitements autorisent des gains supplémentaires qui feront progresser la pratique clinique. Comme les agents actuellement sur le marché maîtrisent l’hypertension pulmonaire par différents mécanismes, il importe d’évaluer leur efficacité relative aux divers stades de la maladie dans le contexte des objectifs du traitement, qui pourraient inclure l’atténuation du processus physiopathologique sous-jacent.
Résumé
L’HTAP est une maladie complexe, hétérogène et multifactorielle. L’HTAP demeure progressive et incurable, mais nous avons à tout le moins des données prouvant indirectement que les traitements actuels améliorent l’issue clinique. Même si tous les traitements actuels ont en commun d’être bénéfiques en diminuant la sévérité de l’hypertension pulmonaire et de l’insuffisance VD, ils y parviennent en empruntant différentes voies. Il importe donc d’envisager les effets relatifs des agents actuels sur les paramètres cliniques ou de substitution de la progression. Les efforts visant à cerner les effets bénéfiques relatifs sur la physiopathologie de l’HTAP ne diminuent pas, à court terme, l’importance d’une meilleure qualité de vie et d’un impact moindre sur les AVQ, surtout aux stades avancés de la maladie, mais la modification des plans d’essais cliniques de façon à explorer des paramètres au-delà de la stabilisation de la capacité fonctionnelle pourrait nous amener à mettre au point des traitements capables d’infléchir le cours naturel de l’HTAP.
Références
1. D’Alonzo GE, Barst RJ, Ayres SM et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343-9.
2. Galiè N, Manes A, Negro L, Palazzini M, Bacchi- Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J 2009;30:394-403.
3. Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. European Respiratory Journal 2010;35: 1079-87.
4. McGoon MD, Kane GC. Pulmonary hypertension: diagnosis and management. Mayo Clinic proceedings Mayo Clinic 2009;84:191-207.
5. Barst RJ, Rubin LJ, Long WA et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334:296-301.
6. McLaughlin VV, Badesch DB, Delcroix M et al. End points and clinical trial design in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54:S97-107.
7. Pulido T, Adzerikho I, Channick RN et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809-18.
8. Barst RJ, Chung L, Zamanian RT, Turner M, McGoon MD. Functional class improvement and 3-year survival outcomes in patients with pulmonary arterial hypertension in the REVEAL Registry. Chest 2013;144:160-8.
9. Galiè N, Hoeper MM, Humbert M et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493-537.