Comptes rendus
Syndromes myéloprolifératifs : de grandes avancées cliniques grâce aux thérapies ciblées
Le présent compte rendu est fondé sur des données médicales présentées lors d'un congrès de médecine reconnu ou publiées dans une revue avec comité de lecture ou dans un commentaire signé par un professionnel de la santé reconnu. La matière abordée dans ce compte rendu s'adresse uniquement aux professionnels de la santé reconnus du Canada.
FRONTIÈRES MÉDICALES - 53e Assemblée/Exposition annuelle de l’American Society of Hematology
San Diego, Californie / 10-13 décembre 2011
San Diego - À mesure que s’enrichissent nos connaissances sur la physiopathologie moléculaire des syndromes myéloprolifératifs (SMP), des agents vraisemblablement capables d’inhiber des étapes clés du processus morbide voient le jour. Dans la myélofibrose, la polycythémie vraie et la thrombopénie essentielle, il a été établi que la voie des janus kinases (JAK) jouait un rôle clé et qu’elle constituait une cible séduisante pour la maîtrise de la maladie. Bien que d’autres agents, y compris des immunomodulateurs, demeurent à l’étude, la découverte des avantages éventuels de l’inhibition d’un médiateur central de l’expression clinique des SMP a accéléré l’évaluation de plusieurs inhibiteurs de JAK, dont un qui a fait l’objet de deux essais de phase III simultanés. Les essais cliniques réalisés à ce jour n’avaient pas la puissance statistique nécessaire pour objectiver un gain de survie, mais ils montrent une régression marquée des symptômes.
Rédactrice médicals en chef : Dre Léna Coïc, Montréal, Québec
Les syndromes myéloprolifératifs (SMP) – notamment la myélofibrose (MF), la polycythémie vraie (PV), la thrombopénie essentielle (TE), la leucémie myéloïde chronique, la leucémie chronique à neutrophiles, la leucémie chronique à éosinophiles et la leucémie à mastocytes – se développent quand une anomalie de la lignée myéloïde accélère l’hématopoïèse, ce qui entraîne l’apparition d’une fibrose dans la moelle osseuse, une surproduction d’hématies et la synthèse de plaquettes dysfonctionnelles. Tous ces syndromes peuvent entraîner une splénomégalie, un risque accru de thrombose et l’apparition de symptômes invalidants. Les mutations des janus kinases (JAK) isolées dans la MF, la TE et la PV sont très répandues, leur fréquence variant entre 50 % (MF et TE) et 95 % (PV) des cas. La dérégulation fonctionnelle des JAK peut survenir en l’absence de mutations pour diverses raisons, par exemple une anomalie de la boucle d’activation du domaine catalytique.
Au vu des nouvelles données indiquant que les inhibiteurs de JAK exercent des effets bénéfiques dans plusieurs SMP, même en l’absence de mutations de JAK, l’inhibition ciblée de la voie JAK-STAT semble importante. La majorité des cas de MF, de PV et de TE, tant primaires que secondaires, se caractérisent par la mutation V617F connue pour entraîner un gain de fonction de JAK2. «Certes, il s’agit d’une cible potentiellement importante du traitement, mais nous savons également que le type sauvage de JAK2 joue un rôle clé dans l’hématopoïèse normale. Il pourrait donc être important d’inhiber JAK2, mais il faut le faire sélectivement afin de ne pas nuire au fonctionnement normal de la moelle», explique le Dr Srdan Verstovsek, Département des leucémies, M.D. Anderson Cancer Center, University of Texas, Houston.
La voie de signalisation JAK s’est avérée une cible prometteuse non seulement dans les syndromes myéloprolifératifs, mais aussi dans un certain nombre de processus prolifératifs comme les cancers et de processus auto-immuns au cours de maladies inflammatoires comme la polyarthrite rhumatoïde. Plusieurs inhibiteurs de JAK sont donc à l’étude dans différentes indications cliniques.
Études COMFORT
Les données de phase III qui ont récemment amené la FDA à homologuer un inhibiteur de JAK dans la MF ont été dévoilées au congrès 2011 de l’American Society of Clinical Oncology (ASCO), puis mises à jour au congrès 2011 de l’American Society of Hematology (ASH). L’agent étudié, le ruxolitinib, a fait l’objet de deux essais cliniques menés simultanément, COMFORT-I et COMFORT-II.
«La constance de l’effet dans tous les sous-groupes est le résultat le plus important des analyses récentes des essais COMFORT. L’avantage relatif associé au ruxolitinib par rapport au meilleur traitement disponible a été observé sans égard au syndrome myéloprolifératif, à la présence ou à l’absence de la mutation V617F, à l’âge (≤65 ou >65 ans) et à la stratification en fonction du volume de la rate», explique la Dre Claire N. Harrison, Département d’hématologie, Guy’s and St. Thomas NHS Foundation Trust, Londres, Royaume-Uni (Figure 1). Aux yeux des chercheurs, le meilleur traitement disponible désignait l’hydroxyurée et divers agents utilisés dans le traitement de l’anémie, comme la prednisone à faible dose, les agents qui stimulent l’érythropoïèse ou les androgènes, explique-t-elle.
Figure 1. COMFORT-II : Efficacité par sous-groupe
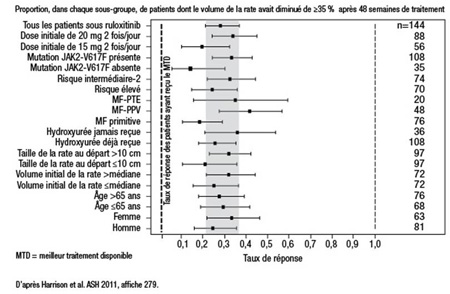
De plan similaire, les deux essais COMFORT regroupaient des patients atteints de MF, de MF post-PV (MF-PPV) ou de MF post-TE (MF-PTE), à risque intermédiaire ou élevé dans tous les cas. Dans le cadre de l’essai COMFORT-I, auquel participaient des centres du Canada, 309 patients ont été randomisés de façon à recevoir par voie orale du ruxolitinib à raison de 15 ou 20 mg
2 fois/jour, selon leur nombre de plaquettes, ou un placebo. Lors de l’essai COMFORT-II, qui s’est déroulé en Europe, 219 patients ont été randomisés de façon à recevoir l’inhibiteur de JAK2 à l’une ou l’autre des posologies selon leur nombre de plaquettes ou le meilleur traitement disponible, qui pouvait inclure de l’hydroxyurée, des transfusions sanguines, des corticostéroïdes, de l’allopurinol et d’autres traitements souvent utilisés dans les soins de soutien.
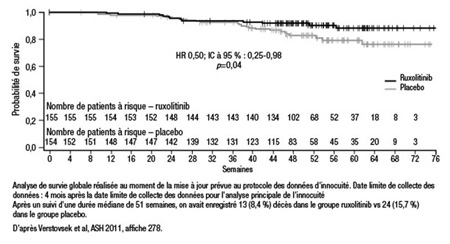
Les résultats des deux essais se renforcent mutuellement. Après 24 semaines de traitement, on a observé une réduction de ≥35 % du volume de la rate chez 42 % des sujets de COMFORT-I vs 1 % des témoins sous placebo (p<0,0001). Dans le cadre de COMFORT-II, toujours après 24 semaines de traitement, les proportions se chiffraient à 31,9 % dans le groupe de traitement actif et à 0 % dans le groupe du meilleur traitement disponible (p<0,0001). Au chapitre du score total de symptômes (malaises abdominaux, satiété précoce, prurit, sueurs nocturnes et douleur osseuse), la proportion de sujets de COMFORT-I chez qui on a observé une diminution de ≥50 % était aussi significativement plus marquée dans le groupe ruxolitinib (45,9 % vs 5,3 %; p<0,0001). Bien que la puissance des essais COMFORT n’ait pas été suffisante pour objectiver une différence entre les groupes sur le plan de la survie dans COMFORT-I, l’inhibition de JAK a augmenté de 50 % la probabilité relative de survie (HR 0,50; IC à 95 % : 0,25-0,98; p=0,04) (Figure 2).
Figure 2. COMFORT-I : Mise à jour de la survie globale (intention de traiter)
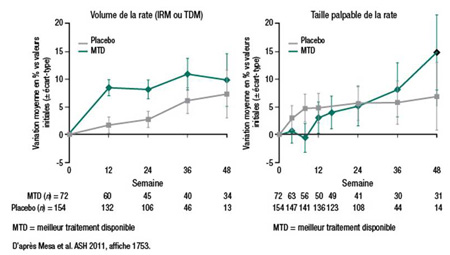
Une analyse post hoc des essais COMFORT a apporté plus de détails sur l’importance relative d’un traitement efficace de la MF. Au chapitre de l’issue clinique, on n’a observé aucune différence entre les témoins de COMFORT-II qui recevaient le meilleur traitement disponible et les témoins de COMFORT-I qui recevaient un placebo, explique le Dr Ruben Mesa, Mayo Clinic, Scottsdale, Arizona (Figure 3). En fait, «dans ces deux groupes, il n’y a eu aucune amélioration cliniquement significative entre le début et la fin de l’étude quant aux scores de symptômes ou de qualité de vie», et l’augmentation du volume de la rate était «similaire numériquement parlant». En effet, les données semblent indiquer qu’avant l’avènement du ruloxitinib, le meilleur traitement disponible était associé à «une amélioration négligeable du volume de la rate, des symptômes ou de la qualité de vie».
Figure 3. Études COMFORT :
Variation moyenne du volume de la rate au fil du temps, vs volume initial
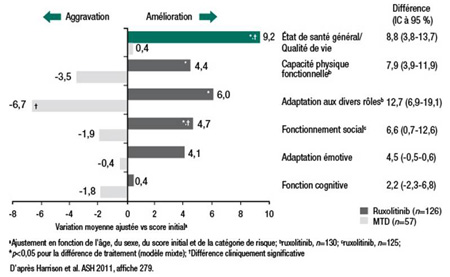
Le protocole de l’essai COMFORT-II prévoyait une analyse rigoureuse de la qualité de vie. Au cours de l’essai, en effet, les patients devaient répondre régulièrement aux questionnaires QLQ-C30 et FACT-Lym, tous deux mis au point par l’Organisation européenne pour la recherche et le traitement du cancer (OERTC). Le questionnaire QLQ-C30 évalue 5 aspects fonctionnels et 9 symptômes. Quant au questionnaire FACT-Lym, il est conçu pour évaluer 4 aspects du bien-être et permet notamment de suivre l’évolution de l’état émotionnel et social. Ces questionnaires ont objectivé une détérioration de la qualité de vie chez de nombreux patients qui recevaient le meilleur traitement disponible, alors que le ruloxitinib a été associé à une amélioration constante qui était non seulement statistiquement significative, mais aussi «cliniquement importante», précise la Dre Harrison (Figure 4).
Figure 4. Variation moyenne ajustée du score de qualité de vie globale et des scores fonctionnels, vs scores initiaux, selon le questionnaire QLQ-30C de l’OERTC
Données à l’appui de l’inhibition de JAK2
Le bénéfice associé à l’inhibition de JAK2 que l’on a observé dans les essais COMFORT, même en l’absence de la mutation caractéristique V617F, valide le rôle clé de la voie JAK2 dans les maladies étudiées et en tant que cible primordiale du traitement. Il n’est donc pas étonnant que d’autres inhibiteurs de JAK2 soient en développement. Des essais cliniques portant sur plusieurs agents, entre autres un essai de phase II sur le pacritinib, ont été présentés au congrès. Dans l’ensemble, ces essais sont venus ajouter du poids à la pertinence de l’inhibition de JAK2 dans ces maladies. Dans le cadre d’un essai de phase II non comparatif sur le pacritinib, qui visait surtout à évaluer la variation du volume de la rate et l’innocuité du traitement, 34 patients atteints de MF et de splénomégalie ont reçu 400 mg/jour comme dose initiale.
«Nous avons noté une diminution de la splénomégalie palpable chez près de 90 % des patients, une diminution ≥50 %
chez 44 % des patients et une régression clinique chez 18 % des sujets», souligne le Dr Rami S. Komrokji, Moffitt Cancer Center, Tampa, Floride. Fait digne de mention, la diminution du volume de la rate était comparable chez tous les patients, que le nombre de plaquettes ait été faible ou normal. Après 6 mois de suivi, la diminution du volume de la rate était corrélée avec une diminution significative des symptômes de la MF, notamment les douleurs abdominales, les douleurs osseuses et la fatigue. Les effets indésirables étaient de nature digestive pour la plupart, mais ils étaient de faible intensité et pouvaient être traités, poursuit le Dr Komrokji.
«Dans le traitement de la splénomégalie et des symptômes constitutionnels associés à la MF, le pacritinib offre une efficacité prometteuse à une dose qui n’entraîne qu’une myélosuppression minime, ajoute-t-il, faisant en cela allusion aux craintes que soulève l’inhibition de JAK2 et qui ne se sont pas encore matérialisées dans les essais cliniques menés à ce jour. «Du fait qu’il n’entraîne pas de myélosuppression, cet inhibiteur de JAK2 revêt une importance particulière pour les patients atteints de MF dont l’hématopoïèse est altérée.»
Stratégies prometteuses dans les SMP
Cela dit, les inhibiteurs de JAK2 ne sont pas les seuls traitements prometteurs dans les SMP. Plusieurs séries de nouvelles données ont été présentées sur les immunomodulateurs, les inhibiteurs des histone déacétylases (HDAC) et l’interféron alfa-2a pégylé. Dans le cadre d’un résumé de trois essais consécutifs sur le pomalidomide, un immunomodulateur, on a présenté des données sur 82 patients, dont 63 avaient été évalués lors d’un essai de phase III. Au départ, les patients dépendaient tous de transfusions de concentrés érythrocytaires. Même si seulement 22 (27 %) répondaient aux critères de réponse prédéfinis de régression de l’anémie, l’état clinique de 21 (96 %) répondeurs s’était amélioré en quelques mois de traitement.
Les deux meilleurs prédicteurs de la réponse étaient la présence de la mutation JAK2-V617F et l’absence de splénomégalie, explique le Dr Kebede Begna, Mayo Clinic, Rochester, New York. Si la survenue de neuropathies périphériques fait parfois obstacle au traitement à long terme, certains patients ont pu demeurer sous pomalidomide jusqu’à 36 mois.
Le givinostat, inhibiteur des HDAC qui cible les cellules caractérisées par la mutation JAK2-V617F, a été évalué chez 44 patients atteints de PV qui ne répondaient pas à la dose maximale tolérée d’hydroxyurée. Dans un essai de phase II, les patients demeuraient sous hydroxyurée et étaient randomisés de façon à recevoir du givinostat à 50 ou 100 mg. La régression des symptômes, qui ne différait pas de façon marquée entre les deux groupes, a été observée chez 95 % des patients. Le traitement a été particulièrement efficace pour soulager le prurit, affirme le Dr Guido Finazzi, Ospedali Riuniti di Bergamo, Italie. Tous les effets indésirables liés au traitement, y compris la diarrhée et la thrombopénie, étaient de grade 2 ou inférieur, de sorte que le développement de cet agent vaut la peine d’être poursuivi, surtout chez les patients présentant un prurit réfractaire.
Plusieurs études sur l’interféron alfa-2a pégylé ont été présentées au congrès, dont une à laquelle plusieurs établissements ont participé. Dans le cadre de cette étude rétrospective, les taux de réponse ont été évalués chez 115 patients, dont 54 (47 %)
souffraient de PV, 44 (38 %) de TE et 17 (15 %) de MF. La majorité des patients avaient déjà reçu au moins un traitement, notamment l’hydroxyurée, l’anagrélide, l’AAS, un interféron non pégylé ou des saignées. La dose initiale médiane se chiffrait à 45 g
(22,5 à 100 g).
Les taux de réponse étaient élevés dans les trois sous-types de SMP, et environ la moitié des patients atteints de PV et de TE ont obtenu une réponse complète. Les signes de toxicité, tant hématologique que non hématologique, étaient généralement légers, précise l’auteure principale, la Dre Krisstina Gowin, Keck School of Medicine, University of Southern California, Los Angeles.
Bien que 17 % des patients aient abandonné le traitement pour cause de toxicité, le traitement a été qualifié de «bien toléré», compte tenu, en particulier, de l’activité du traitement contre les symptômes des SMP. «Comme le traitement cytoréducteur avait échoué chez la majorité des patients, ces résultats étayent l’efficacité de l’interféron alfa-2a pégylé rapportée antérieurement dans les SMP», poursuit la Dre Gowin.
Résumé
Les résultats d’une étude de phase III sur un inhibiteur de JAK – notamment une diminution substantielle du volume de la rate et une impressionnante régression des symptômes – témoignent des progrès que l’on réalise dans le traitement des SMP. Cet essai a d’ailleurs mené à l’homologation de cet agent dans le traitement de la MF aux États-Unis. D’autres inhibiteurs de JAK sont en développement, et des traitements dotés d’autres modes d’action pourraient élargir l’éventail d’options dans le traitement de ces maladies. L’activité que l’on observe dans ce domaine est fort encourageante vu le nombre restreint d’options de traitement à ce jour.
Questions et réponses
Les questions et réponses qui suivent sont tirées d’un entretien avec la Dre Claire N. Harrison, Département d’hématologie, Guy’s and St. Thomas NHS Foundation Trust, Londres, Royaume-Uni.
Q : Aux États-Unis, la FDA a homologué le ruxolitinib dans le traitement des SMP sur la foi des essais COMFORT. Comment pensez-vous utiliser cet agent dans la pratique?
R : Je serais portée, je crois, à le prescrire à tous mes patients atteints de MF symptomatique. La régression des symptômes dans les essais COMFORT m’a vraiment impressionnée. À vrai dire, la recul de ces symptômes très débilitants a été spectaculaire. Je serais même tentée de prescrire du ruxolitinib dans les cas de SMP à risque intermédiaire, pour autant que les patients aient une splénomégalie et des symptômes.
Q : A-t-il été nécessaire de diminuer la dose ou d’apporter d’autres modifications au traitement en raison d’effets indésirables chez certains patients?
R : Il n’y a eu aucun effet indésirable grave dans notre essai. Dans certains cas, selon la dose administrée, nous avons observé une réapparition des symptômes de MF, mais aucun effet indésirable n’a nécessité de diminution de la dose. Au début de l’étude, la dose de 20 mg a été associée à un taux de réponse plus élevé que la dose de 15 mg, mais au terme des 48 semaines, il n’y avait aucune différence, et la dose plus forte n’a pas semblé associée à une hausse importante du risque d’effets indésirables.
Q : Le ruxolitinib est-il associé à un gain de survie?
R : COMFORT-II n’avait pas la puissance statistique voulue pour évaluer la survie, mais COMFORT-I a objectivé un gain de survie significatif par rapport au placebo. La poursuite du traitement nous permettra avec le temps d’accumuler des données sur l’issue clinique à long terme. Le profil de tolérabilité donne tout lieu de croire que le traitement à long terme est une réelle possibilité, mais nous avons besoin d’un suivi plus long pour mieux évaluer l’innocuité, le risque de résistance et la possibilité d’une maîtrise indéfinie de la maladie.